Как и в случае гомогенной химической реакции, константа гетерогенного равновесия равна отношению произведения равновесных концентраций (активностей) или парциальных давлений продуктов реакций к произведению равновесных концентраций (активностей) или парциальных давлений исходных веществ в степенях, равных стехиометрическим коэффициентам в уравнении. Для реакции пароводяной конверсии углерода константа равновесия имеет вид: К р = (р СО2)р (р Н2)2р/(р Н2О)2р, для восстановления металла
К р = (р Н2O)р/(р Н2)р.
Из приведенных выражений следует, что в уравнения констант гетерогенного химического равновесия не входят концентрации твердых веществ, участвующих в прямой и обратной реакциях. Это особенность гетерогенного химического равновесия. Так как прямая и обратная реакции протекают на одной и той же поверхности раздела фаз, то площадь поверхности раздела фаз также не входит в уравнение константы химического равновесия. Константа гетерогенного химического равновесия зависит от температуры. Она возрастает с увеличением температуры для эндотермической прямой реакции и уменьшается с увеличением температуры в случае экзотермической прямой реакции. Расчеты проводятся по тем же формулам, что и для гомогенных реакций. Смещение равновесия гетерогенных реакций подчиняется принципу Ле Шателье. При повышении температуры оно смещается в сторону эндотермической реакции. При повышении давления или концентрации исходных веществ равновесие смещается в сторону образования продуктов реакции, при повышении концентрации или давления продуктов реакции равновесие смещается в сторону обратной реакции. При повышении общего давления равновесие сдвигается в направлении уменьшения числа молекул газообразных веществ. Твердые исходные вещества и продукты реакции не влияют на смещение гетерогенного химического равновесия.
39) Реакции комплексообразования. Типы и св-ва комплексных соединений. Константа устойчивости. Использование комплексных соединений для концентрирования, обнаружения лекарственных в-в.
Примеры компл. соед-й: хлорофилл – комплекс магния с порфиринами, гемоглобин – компл. Fe2+ с порфириновыми циклами, инсулин – комплекс цинка, вит. В12 - комплекс кобальта, платинол – компл. платины и т.д. Роль компл. соед-й: на основе фармакологически активных комплексных соед-й мб понизить токсичность как металла, так и лигандов, связанных в комплексе. Напр., ядовитый цианид калия теряет свою токсичность при связывании в ферроцианид (желтая кровяная соль) или в феррицианид (красная кровяная соль).
1. цианид калия переводит Fe2+ в белый осадок гексацианоферрата(II) железа(II) (а не в цианид железа(II), как считалось ранее, что вытекает из взаимодействия этого цианида со щёлочью: 3Fe(CN)2 + 4KOH → 2Fe(OH)2↓ + K4[Fe(CN)6]):3Fe2+ + 6CN− → Fe2[Fe(CN)6]↓
2. затем осадок растворяется в избытке KCN с образованием «жёлтой кровяной соли»:Fe2[Fe(CN)6] + 12CN− → 3[Fe(CN)6]4−
3. Из гексацианоферрата(III) калия можно получить гексацианоферрат(II) калия с помощью перекиси водорода в щелочной среде {\displaystyle {\mathsf {2K_{3}[Fe(CN)_{6}]+H_{2}O_{2}+2KOH{\xrightarrow {}}\ 2K_{4}[Fe(CN)_{6}]+2H_{2}O+O_{2}{\uparrow }}}}(но в нейтральной среде эта реакция протекает в обратную сторону).
Состав: металл-комплексообразователь (М), с кот. связаны лиганды (L). Атом Mи лиганды L образуют внутреннюю сферу комплекса (при написании формулы – [заключ. в квадратн. скобки]). Лигандами мб нейтральные молекулы (обычно основного хар-ка), отрицат. заряж. анионы (ацидогруппы). Простые положит. заряж. группы в роли лигандов не выступ. Если внутрення сфера комплекса несет отрицат./положит. заряд, то для компенсации этого заряда необх. ионы, образующие внешнюю сферу (это мб не только ионы, но и нейтральн. молекулы – вода, в т.ч. кристаллизационная).


Лиганд образует с металлом-комплексообразователем координационную связь различн. химич. природы (ионная, ковалентная, полярная); мб одинарной, двойной, тройной.
Координационное число центрального атома металла – это число координац. связей, образуемых атомом металла-комплексообразователя с лигандами (мб иметь значения от 2 до 12 включительно).
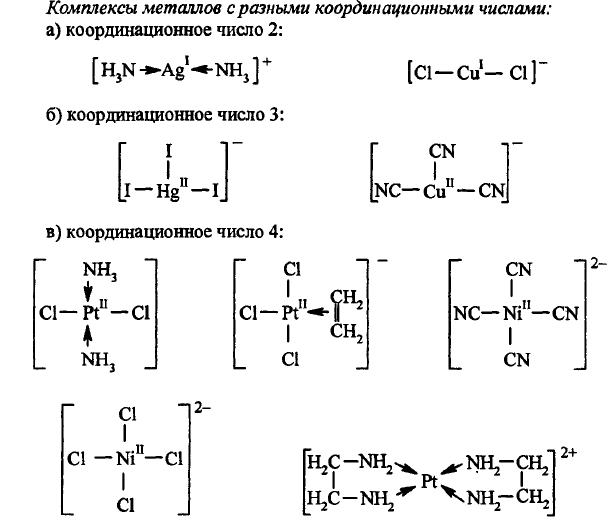
Дентантность – число координационных связей, образуемых одним и тем желигандом с одним атомом металла-комплексообразователя (типы лигандов - мб монодентантными и полидентантными).
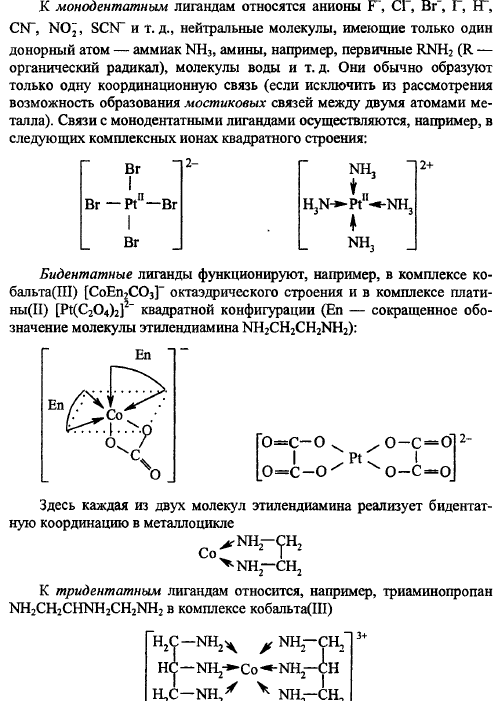
Комплексные соед-я мб катионного, анионного типа и комплексами-неэлектролитами (точнее слабыми электролитами):

Если комплекс содерж. только один металл-комплексообразователь, то он назыв. одноядерным, если 2 и более – многоядерным (типы).

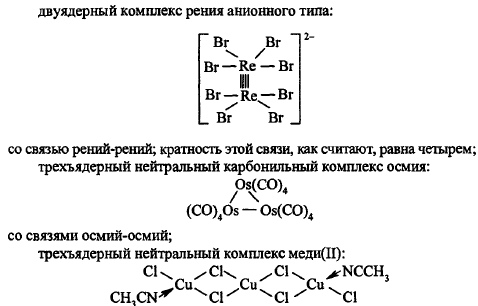
Если полиядерные комплексы содержат атомы Ме одинаковой хим. природы, то они назыв. (типы) гомометаллическими; если разной – гетерометаллической:
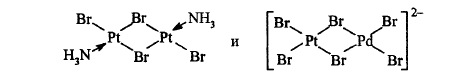
Если лиганд связан в полиядерном компл. одновременно с двумя-тремя атомами Ме, то его назыв. мостиковым; если больше трёх атомов Ме – кластерные соединения (тип):
Константа устойчивости:




Использование комплексных соединений для концентрирования, обнаружения лекарственных в-в: 


40) Окислительно-восстановительные реакции в аналитической химии. Обратимые и необратимые окислительно-восстановит. системы и их потенциалы. Уравнение Нернста. Стандартный и реальный окислительно-восстановительные потенциалы.
Отличительным признаком окислительно-восстановительных (редокс-) реакций является перенос электронов между реагирующими частицами – ионами, атомами, молекулами, комплексами, в результате чего изменяется степень окисления реагирующих частиц, например, Fe2+ - e- → Fe3+. Поскольку электроны не могут накапливаться в растворе, одновременно должны протекать два процесса – отдача и принятие электронов, т. е. процесс окисления одних и восстановления других частиц. Таким образом, любая окислительно-восстановительная реакция всегда может быть представлена в виде двух полуреакций: a Оx1 + b Red2 → а Red1 + b Оx2, а Оx1 + n е- → a Red1, b Red2 – n е- → b Оx2, где Ox – окисленная форма, Red – восстановленная форма.
Окислительно-восстановительные реакции, как и все динамические процессы, в той или иной мере обратимы. Направление реакций определяется соотношением электроно-донорных свойств компонентов системы одной окислительно-восстановительной полуреакции и электроно-акцепторных свойств второй (при условии постоянства факторов, влияющих на смещение равновесных химических реакций). Перемещение электронов в ходе окислительно-восстановительных реакций приводит к возникновению потенциала. Таким образом, потенциал, измеряемый в вольтах, служит мерой окислительно-восстановительной способности соединения.

Редокс-пара – это система из окисленной и восстановленной форм данного в-ва, в кот. окисленная форма (ок-ль) является акцептором электронов и восстанавливается, принимая электроны, а восстановленная форма выступает в роли донора электронов и окисляется, отдавая электроны.
Редокс-амфотерные соед-я – в-ва, кот. в одних р-циях мб окислителями,а в др. – восстановителями (Н2О2; NO2-):

Эффективность окислительных или восстановительных св-в данного в-ва зависит от его природы, от условий протекания окислительно-восстановительной реакции и определяется величиной электродного потенциала редокс-пары (окислительно-восстановительного потенциала редокс-пары). Этот потенциал экспериментально определяют с помощью окислительно-восстановительного электрода.
Окислительно-восстановительный электрод – это электрод, состоящий из инертного материала (металлические платина, золото, вольфрам, титан, графит), погруженного в водный р-р, в кот. имеются окисленная и восстановленная формы данного в-ва. Применяются две разновидности ОВ электродов: потенциал которых не зависит от активности ионов водорода, и эектроды, потенциал которых зависит от активности ионов водорода.



ОВ потенциалы, как и др. электродные потенциалы, принято отсчитывать от потенциала стандартного водородного электрода, кот. условно приним. равным нулю. Потенциалы, отсчит. от потенциала стандартного водородного электрода, назыв. условными/относительными потенциалами по водородной шкале.

Стандартный водородный электрод не явл. ОВ, а относится к электродам первого порядка, потенциал кот. зависит от активности соответствующих катионов (в данн. случ. – катинов водорода).
Условный/относительный ОВ потенциал редокс-пары (электродный потенциал редокс-пары) – это электродвижущая сила (ЭДС) гальванической цепи, составленной из данного ОВ электрода и стандартного водородного электрода.

Если в ОВ реакции не участв. ионы водорода, то реальный условный ОВ потенциал Е редокс-пары опис. ур-ем НЕРНСТА:


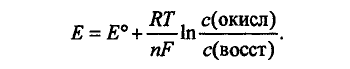 (6.2)
(6.2)
Активности заменяют на конц-ии для сильно разбавленных р-ров (когда коэфф. активности равны единице и численно совпадают с конц-ей; также мб заменить, когда р-ры не сильно разб., но коэфф. окисл. и восстановл. форм приблизит. одинаковы из-за близости химич. природы обеих форм (напр., для хинона и гидрохинона).




Направление протекания реакций:



41) Инструментальные методы анализа. Классификация и область применения в фармации и токсикологии.
Общая характеристика инструментальных (физико-химических) методов:




42) Спектроскопические методы анализа. Спектр электромагнитного излучения. Энергия фотонов, длина волны, частота, волновое число; связь между ними; термины, символы, единицы измерения. Составляющие внутренней энергии частиц и соответствующие им диапазоны электромагнитного излучения.
Имеется целый ряд типов электромагнитного излучения, начиная с радиоволн и заканчивая гамма-лучами. Электромагнитные лучи всех типов распространяются в вакууме со скоростью света и отличаются друг от друга только длинами волн:

На рисунке видно, что энергия излучения уменьшается слева направо, т.е. при увеличении длины волны и уменьшении её частоты. 
Классификация и основные принципы спектроскопических методов: все спектроскопические методы основаны на взаимодействии атомов, молекул или ионов, входящих в состав анализируемого вещества, с электромагнитным излучением. Это взаимодействие проявляется в поглощении или испускании фотонов (квантов). В зависимости от характера взаимодействия пробы с электромагнитным излучением выделяют две группы методов - эмиссионные иабсорбционные. В зависимости от того, какие частицы формируют аналитический сигнал, различают методы атомной спектроскопии и методы молекулярной спектроскопии.
1. По решаемым задачам:
- элементный, когда устанавливается состав пробы по элементам;
- изотопный, когда устанавливается состав пробы по изотопам;
- молекулярный, когда устанавливается молекулярный состав пробы;
- структурный, когда устанавливаются все; или основные структурные составляющие молекулярного соединения.
2. По применяемым методам:
- эмиссионный, использующий спектры излучения, главным образом атомов. Однако возможен эмиссионный анализ и молекулярного состава - в случае определения состава радикалов в газовом разряде. Особым случаем эмиссионного анализа является люминесцентный анализ;
- абсорбционный, использующий спектры поглощения, главным образом молекул и их структурных частей; возможен анализ по спектрам поглощения атомов;
- комбинационный, использующий спектры комбинационного рассеяния твердых, жидких и газообразных проб, возбуждаемые монохроматическим излучением, обычно — светом отдельных линий ртутной лампы;
- люминесцентный, использующий спектры люминесценции вещества, возбуждаемые главным образом ультрафиолетовым излучением или катодными лучами;
- рентгеновский, использующий а)рентгеновские спектры атомов, получающиеся при переходах внутренних электронов в атомах, б) дифракцию рентгеновых лучей при прохождении их через исследуемый объект для изучения структуры вещества;
- радиоспектроскопический, использующий спектры поглощения молекул в микроволновом участке спектра с длинами волн больше 1 мм.
3. По характеру получаемых результатов:
1) качественный, когда в результате анализа определяется состав без указания на количественное соотношение компонентов или дается оценка — много, мало, очень мало, следы;
2) полуколичественный, грубоколичественный или приближенный. В этом случае результат выдается в виде оценки содержания компонентов в некоторых более или менее узких интервалах концентраций в зависимости от применяемого метода приближенной количественной оценки. Этот метод благодаря быстроте нашел широкое применение при решении задач, не требующих точного количественного определения, например, при
сортировке металла, при оценке содержания геологических проб при поисках полезных ископаемых;
3) количественный, при котором выдается точное количественное содержание определяемых элементов или соединений в пробе.
Все эти типы анализа, за исключением качественных, используют упрощенные или точные методы фотометрирования спектров.
4. По способу регистрации спектров различаются следующие методы:
1. Визуальные при наблюдении спектров в видимой области с помощью простых или специализированных спектроскопов (стилоскоп, стилометр). В ультрафиолетовой области возможно наблюдение сравнительно ярких спектров с помощью флуоресцирующих экранов, располагаемых вместо фотографической пластинки в кварцевых спектрографах. Применение электронно-оптических преобразователей позволяет визуально наблюдать спектры в ультрафиолетовой и ближней инфракрасной областях (до 12000А).
2. Фотографические, использующие фотографическую пластинку или пленку для регистрации спектров с последующей обработкой.
3. Фотоэлектрические для ультрафиолетовой, видимой и ближней инфракрасной областей, использующие фотоэлементы разных типов - фотоумножители и фотосопротивления (инфракрасная область). Фотоэлектрические методы иногда называются методами прямого анализа, т.е. анализа без посредства фотографической пластинки.
4. Термоэлектрические для инфракрасной области, в том числе далекой, с использованием термоэлементов, болометров и других типов термоэлектрических приемников.
Рассмотренные выше типы спектрального анализа имеют ряд общих черт, поскольку все они используют спектры атомов или молекул как средство для проведения анализа. Действительно, во всех случаях необходимо в первую очередь получить спектр пробы, затем расшифровать этот спектр по таблицам или атласам спектров, т.е. найти в этом спектре линии или полосы, характерные для определяемых атомов, молекул или структурных элементов молекул. Этим ограничивается качественный анализ. Для получения количественной величины концентрации надо, кроме того, определить интенсивность этих характерных линий или полос (фотометрировать спектр), затем определить величину концентрации, используя зависимость между концентрацией и интенсивностью линий или полос. Зависимость должна быть получена либо на основании теоретических соображений, либо эмпирическим путем в виде аналитической кривой, построенной на основе набора проб с заданными концентрациями (эталоны).
Классификация спектроскопических методов: в эмиссионных методаханализируемая проба в результате ее возбуждения излучает фотоны (кванты). Важнейшие эмиссионные методы - атомно-эмиссионный спектральный анализ (АЭС) и люминесцентный анализ.
В абсорбционных методахизлучениепостороннего источника пропускают через пробу, при этом часть квантов избирательно поглощается атомами или молекулами. Важнейшие методы этой группы - атомно-абсорбционный анализ (ААС) и молекулярно-абсорбционная спектроскопия растворов. Последний метод обычно называют спектрофотометрией или фотометрическим анализом. Абсорбционные методы, как и эмиссионные, используют и для обнаружения, и для количественного определения веществ.
Области электромагнитного спектра, применяемые в химическом анализе: э лектромагнитное излучение может проявляться в разных формах: видимый свет, ультрафиолетовое излучение, инфракрасное (тепловое) излучение, радиоволны, рентгеновские лучи, гамма-лучи. Если все фотоны (кванты) данного излучения имеют одну и ту же энергию, излучение называют монохроматическим, если их энергии различны – полихроматическим. Монохроматический свет имеет определенную длину волны. Полихроматическое излучение характеризуется интервалом длин волн, в который входят все компоненты данного излучения. Более полная характеристика полихроматического излучения – спектр - он показывает распределение интенсивности излучения по длинам волн (или по энергиям, или по частотам).
Электромагнитное изучение охватывает очень широкий интервал длин волн и энергий. Видимый свет соответствует лишь малой части этого интервала. В химическом анализе применяют не все виды излучений. Широко используют «оптический диапазон», границы которого - от 200 нм до 40 000 нм. В этот диапазон входят три области: ультрафиолетовая область (УФ) – 200-400 нм; видимая область – 400-800 нм; инфракрасная область (ИК) - от 800 до 40 000 нм.
Внутри каждой области иногда выделяют еще более узкие участки, имеющие собственные названия. Так, в ИК-спектрах выделяют особую «область отпечатков пальцев». Название связано с высокой информативностью этой области для опознания индивидуальных органических веществ. В видимой области отдельные участки характеризуют цветом излучения.
Спектры излучения и поглощения: как правило, анализируемая проба излучает и поглощает полихроматический свет, включающий кванты разной энергии и разной длины волны. Однако для аналитика предпочтительнее измерять испускание или поглощение света, в котором все кванты примерно одинаковы по энергии, соответствуют одной длине волны. Чтобы выделить ее из полихроматического излучения, нужно особое устройство – монохроматор (рис.1).
Спектральные приборы, снабженные монохроматорами, называют спектрометрами, спектрографами или стилоскопами, в зависимости от используемого в них приемника излучения, то есть от того, какой способ регистрации спектра (фотоэлектрический, фотографический или визуальный) применяется в этих приборах. С помощью таких приборов можно зарегистрировать спектр излучения или спектр поглощения исследуемой пробы.
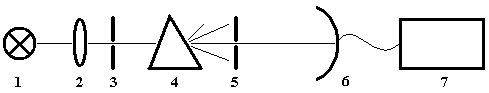
Рис.1. Схема спектрометра с призменным монохроматором
1 – источник света, 2 – фокусирующая оптика, 3 – входная щель, 4 – призма, 5 – выходная щель, 6 – приемник (фотоэлемент), 7 – регистрирующее устройство (микроамперметр и т.п.).
Спектр излучения пробы показывает, на каких длинах волн она преимущественно излучает свет при возбуждении (например, при сильном нагревании). Спектр излучения регистрируют в координатах: интенсивность (I) излучения - длина волны (λ).
Спектр поглощения пробы показывает, на каких длинах волн она преимущественно поглощает излучение внешнего источника. Такие спектры обычно регистрируют в координатах A – λ, где А - количественная характеристика поглощения света на данной длине волны, называемая оптической плотностью.
Спектры поглощения и излучения одного и того же вещества в некоторой области длин волн очень похожи. Чтобы понять, почему это так, надо вспомнить, что атом может находиться только в определенных состояниях, которым отвечают дискретные энергетические уровни. Переходя под воздействием внешнего излучения в более возбужденное состояние, атом должен приобрести дополнительную энергиюза счет поглощения кванта. Поэтому в спектре поглощения пробы на длине волны, соответствующей энергии, появится линия (пик). В атомах данного элемента возможны и другие энергетические переходы (с другими значениями энергии). Все они реализуются одновременно, приводя к другим линиям в спектре поглощения данного элемента.Теперь рассмотрим атомы, которые уже переведены в возбужденное состояние, например, под действием высокой температуры. Через короткое время (10-7 – 10-8 сек) после возбуждения они самопроизвольно возвращаются в основное состояние, излучая кванты. Энергии этих квантов равны разностям энергий соответствующих состояний. Каждому переходу соответствует некоторая длина волны, некоторая линия в спектре испускания. Поскольку поглощение и испускание света определяются одними и теми же энергетическими переходами, в спектрах поглощения и излучения данного элемента наблюдаются одни и те же линии.
Особенности спектров разного типа и их аналитическое применение: атомные спектры поглощения и излучения, наблюдаемые во всем оптическом диапазоне, определяются переходами электронов, относящихся к наружным слоям («валентные электроны»). Таким образом, атомные спектры по своей природе являются электронными,а по внешнему виду - линейчатыми. Положение спектральных линий в шкале длин волн и их относительную интенсивность используют как идентификационные признаки в качественном элементном анализе.
Молекулярные спектры излучения или поглощения обычно не являются линейчатыми. Вид молекулярных спектров в разных диапазонах длин волн различен, поскольку различно происхождение соответствующих спектров. Спектры поглощения молекул в видимой или УФ-области являются широкополосными. Они дают сравнительно мало информации для выяснения состава и структуры поглощающих молекул. Это мешает проведению качественного анализа по спектрам в УФ - или видимой области.
Изучение молекулярных спектров –это важнейший способ количественного химического анализа. Спектры нужны для решения сложных задач. А именно:
1) По спектру индивидуального вещества выбирают ту длину волны, на которой в дальнейшем, в ходе количественного анализа, будут измерять аналитический сигнал этого вещества (I или А). Если для определения какого-либо элемента в атомно-эмиссионном спектральном анализе используют наиболее интенсивные линии эталонного спектра, то в молекулярно-абсорбционном (спектрофотометрическом) анализе аналитический сигнал обычно измеряют на длине волны, соответствующей максимуму на спектральной кривой.
2) Сопоставляя спектры предполагаемых компонентов пробы, выясняют возможность определения одних веществ в присутствии других. Если спектры компонентов пробы накладываются друг на друга, результаты анализа смеси будут завышенными. Для снижения систематических погрешностей, связанных с наложением спектров, созданы особые приемы измерений и расчета результатов. Другие выходы из положения - маскирование или предварительное отделение мешающих компонентов.
Атомно-эмиссионный спектральный анализ: давнобыло замечено, что цвет пламени меняется при введении в него некоторых веществ - этот эффект стали использовать в анализе (по окраске пламени различали соду (Na2CO3) и поташ (К2CO3). Позже был установлен линейчатый характер спектров пламени. Спектры начали фотографировать, определять длины волн отдельных линий - по наличию определенных спектральных линий можно судить о присутствии в пробе тех или иных элементов. В результате исследований Бунзена и Кирхгофа был установлен качественный (элементный) состав многих минералов и даже небесных тел; открыт ряд ранее не известных элементов (таллий, индий и др.); созданы первые, еще не очень точные способы количественного спектрального анализа.
Затем для получения спектров стали применять электрическую дугу и искру, это позволило определять те элементы, которые не возбуждаются в пламени. Было доказано, что спектральный анализ применим для обнаружения и определения элементов, независимо от их степени окисления и от того, в составе каких химических соединений они находились в исходной пробе.
Принцип метода: во всех вариантах АЭС пробу вносят в источник возбуждения, где тем или иным способом создается высокая температура (тысячи градусов). Образуется плазма (совокупность возбужденных атомов, ионов и электронов). В ней последовательно проходят следующие процессы: испарение пробы;атомизация первоначальных продуктов испарения (молекул или ионов); возбуждение образовавшихся атомов; испускание света возбужденными атомами.
Возникающее в ходе анализа полихроматическое излучение пробы фокусируют и направляют на входную щель спектрального прибора, где оно разлагается в спектр и регистрируется соответствующим приемником (фотоэлемент, диодная линейка, фотопластинка и др.). Можно наблюдать спектр и визуально (именно так работали Бунзен и Кирхгоф), однако это небезопасно для глаз. Для качественного анализа полученный спектр сопоставляют с эталонными спектрами разных элементов. По одному спектру пробы можно быстро и надежно обнаружить многие, а то и все присутствующие в ней элементы. Для количественногоанализа надоизмерить интенсивность излучения пробы на некоторых заранее выбранных длинахволн. На практике измеряют не саму интенсивность излучения (число квантов, испускаемых пробой в единицу времени), а зависящие от нее другие величины - фототок, абсолютное или относительное почернение фотопластинки и др. По этим аналитическим сигналам и рассчитывают содержания разных элементов, пользуясь градуировочными графиками, заранее полученными с помощью подходящих эталонов.
Приборы для спектрального анализа включают в себя три основных блока: блок возбуждения, диспергирующее устройство и блок регистрации излучения. Разные варианты АЭС различаются по способу возбуждения пробы и по способу регистрации спектра.
Источники возбуждения: в качестве источников возбуждения применяют пламя, электрическую дугу, искру, а также высокочастотную индуктивно-связанную плазму (ИСП, ICP). Три первых источника являются «классическими». Недавно созданный метод ICP только входит в практику работы аналитических лабораторий, но именно он обеспечивает наилучшие результаты. В научных исследованиях используют также импульсный разряд, микроволновой разряд, лазерное излучение и некоторые другие источники плазмы.
Способы регистрации спектра:
1) фотографическая регистрация достаточно проста по технике, доступна. Этот «классический» способ позволяет получать и измерять даже очень слабые сигналы микропримесей. Одновременно регистрируются линии всех компонентов пробы. Сфотографированные спектры можно долго хранить и в любое время провести повторные измерения.
Схема спектрографа похожа на схему спектрометра, но выходная щель и измерительное устройство в данном случае не нужны. После разложения излучения пробы по длинам волн оно направляется на фотопластинку, содержащую в своем поверхностном слое кристаллы бромида серебра. В местах, куда попадет излучение, образуется металлическое серебро. В результате проявления и закрепления фотопластинки ее почернение во много раз усиливается. На пластинке остается спектр пробы в виде ряда черных линий одинаковой высоты и ширины, но с разной степенью почернения (картинка напоминает штрих-код товара). Все эти линии являются фотографиями входной щели, сделанными на разных длинах волн, соответственно спектральному составу излучения пробы. На одну и ту же фотопластинку можно последовательно сфотографировать десятки спектров, размещая их друг под другом. Почернение аналитических линий на проявленной фотопластинке измеряют с помощью вспомогательного прибора – микрофотометра.
2) фотоэлектрическая регистрация основана на применении фотоэффекта - фототок приблизительно пропорционален интенсивности излучения, вызывающего фотоэффект. Фотоэлектрическая регистрация спектров более экспрессна, чем фотографическая. Исключается трудоемкая обработка фотопластинок и последующие измерения почернений, соответственно устраняются погрешности, возникающие на этих стадиях анализа. Приборы для фотоэлектрической регистрации спектров разнообразны.
Качественный анализ: существуют обширные спектральные атласы, где приведены эталонные спектры испускания большого числа элементов с указанием длин волн и относительных интенсивностей линий. Однако не все линии эталонного спектра можно найти в спектре пробы, содержащей данный элемент. По мере уменьшения концентрации компонента в пробе интенсивность излучения уменьшается настолько, что часть линий (наименее интенсивные) уже не регистрируется данным прибором. Последними при разбавлении пробы исчезают так называемые последние линии (как правило, наиболее интенсивные). Для каждого элемента эти линии хорошо известны. Чтобы проверить наличие некоторого элемента в пробе, проверяют наличие в спектре пробы нескольких последних линий этого элемента. Но если в спектре обнаружена линия, длина волны которой численно совпадает с длиной волны последней линии искомого элемента, то это вовсе не означает, что она действительно принадлежит данному элементу. Дело в том, что в спектрах многих элементов насчитывается очень большое число линий (у калия – несколько десятков, у железа – несколько сот, у урана – несколько тысяч). Линии разных элементов часто случайно совпадают по длине волны («межэталонные наложения»). Могут совпадать и последние линии. Поэтому окончательный вывод о присутствии в пробе интересующего элемента следует делать, если в спектре пробы установлено наличие нескольких линий, совпадающих по длине волны с линиями эталонного спектра данного элемента и заведомо свободных от межэталонных наложений. Полезно также проверить наличие характерных комбинаций линий (дублетов, триплетов). Спектры пробы и эталона должны совпадать и по относительной интенсивности разных линий.
Количественный анализ: интенсивность излучения в АЭС определяется концентрацией возбужденных атомов. Если все условия анализа одинаковы, получим прямо пропорциональную зависимость интенсивности излучения на данной длине волны от концентрации элемента в пробе: 
Расчет концентраций ведут по предварительно построенным градуировочным графикам.