Белковая недостаточность - следствие дефицита белка, а также тяжелых заболеваний при достаточном поступлении белка с пищей. Развивается также при однообразном питании белками растительного происхождения. Характеризутся отрицательным азотистым балансом, гипопротеинемией, нарушениями коллоидно-осмотического и водно-солевого обмена.
В развивающихся странах среди детей распространено заболевание квашиоркор. Оно возникает при тяжелых формах пищевых дистрофий. Наблюдаются тяжелые поражения печени, остановка роста, резкое снижение сопротивляемости организма инфекциям, отечность, атония мышц. Болезнь часто заканчивается летальным исходом.
Гипераммониемия – повышение содержания аммиака в крови. Гипераммониемиясопровождается появлением следующих симптомов: тошнота, повторяющаяся рвота; головокружение, судороги; потеря сознания, отек мозга (в тяжёлых случаях); отставание умственного развития (при хронической врождённой форме).
Причинами гипераммониемии могут быть генетические дефекты ферментов орнитинового цикла в печени. Известны пять наследственных заболеваний, обусловленных дефектом пяти ферментов орнитинового цикла (таблица 2). Снижение активности какого-либо фермента синтеза мочевины приводит к накоплению в крови субстрата данного фермента и его предшественников. Например, недостаточность первого фермента цикла - карбамоилфосфатсинтетазы I - привводит к накоплению аммиака и его предшественников - аминокислот; главным образом глутамина и аланина. Концентрация аммиака может превышать нормальную в 10-20 раз, концентрация глутамина - в 2-3 раза.
При циррозе и других заболеваниях печени также часто наблюдают гипераммониемию, т.к. мощность орнитнового цикла снижена.
Точный диагноз типа гипераммониемии устанавливают путем определения метаболитов орнитинового цикла в крови или моче, а также путем измерения активности ферментов цикла в биоптате печени.
Лечение больных с различными дефектами орнитинового цикла в основном направлено на снижение концентрации аммиака в крови за счёт малобелковой диеты, введения кетоаналогов аминокислот в рацион и стимуляцию выведения аммиака в обход нарушенных реакций:
Гипераминоацидурия – увеличение выделения аминокислот с мочой.
Выделяют почечную гипераминоацидурию, связанную с дефектами реабсорбции аминокислот в почках, и внепочечную, обусловленную увеличением концентрации аминокислот в крови.
Часто встречаются наследственные дефекты всасывания аминокислот в почках. Например,причиной цистинурии - выделения с мочой цистина (до 50 раз больше нормы) – является врожденный дефект переносчиков, участвующих в реабсорбции в почках. Уровень цистина в крови при этом обычно не выше нормальных величин. Примером внепочечной гипераминоацидурии может быть аргининосукцинурия.
Повышение уровня мочевины в крови происходит при нарушениях выведения мочевины, связанного с заболеваниями почек и мочевыводящих путей; чрезмерном катаболизме белков либо употреблениии в пищу чрезмерного количества белка (в результате усиленного синтеза мочевины); обезвоживание организма.
Снижение уровня мочевины в крови наблюдается при врожденной недостаточности или отсутствии ферментов, участвующих в орнитиновом цикле мочевинообразования; при тяжелых поражениях печени; при диете с низким содержанием белка, голодании и др.
| Заболевание
| Дефект ф-та
| Тип наследования
| Клинические проявления
| Метаболиты
|
| Кровь
| Моча
|
| Гипераммониемия, тип I
| Карбамоил-фосфат-синтетаза I
| Аутосомно-рецессивный
| В течение 24-48 ч после рождения кома, смерть
| Глн, Ала, NH3
| Оротат
|
| Гипераммониемия, тип II
| Орнитинкарбамоил-трансфераза
| Сцепленный с Х-хромосомой
| Гипотония, снижение толерантности к белкам
| Глн, Ала, NH3
| Оротат
|
| Цитруллинемия
| Аргинино-сукцинат-синтетаза
| Аутосомно-рецессивный
| Гипераммониемия тяжелая у новорожденных. У взрослых - после белковой нагрузки
| Цитруллин, NH3
| Цитруллин
|
| Аргининосукцинатурия
| Аргинино-сукцинатлиаза
| Аутосомно-рецессивный
| Гипераммониемия, атаксия, судороги, выпадение волос
| Аргининосукцинат, NH3
| Аргининосукцинат, Глн, Ала, Лиз
|
| Гипераргининемия
| Аргиназа
| Аутосомнорецессивный
| Гипераргининемия
| Apг, NH3
| Apг, Лиз, орнитин
|
Цистиноз (часто связывают с синдромом Фанкони) — врожденное нарушение реабсорбции почти всех аминокислот (за исключением циклических) в канальцах почек. В результате в 5-10 раз увеличивается экскреция аминокислот, в том числе в 20—30 раз цистина и цистеина. Цистин откладывается в селезенке, почках, коже внутриклеточно накапливаются кристаллы цистина в лизосомах. Нарушается рост, страдает функция почек.
Цистинурия (цистин-лизинурия). При этом наследуемом метаболическом заболевании экскреция цистина с мочой в 20-30 раз превышает норму. Значительно повышается также экскреция лизина, аргинина и орнитина. Цистинурию рассматривают как следствие нарушения процессов транспорта в почках. Поскольку цистин слаборастворим, у больных цистинурией может происходить образование цистиновых камней в почечных канальцах
Алкаптонурия обусловлена недостаточностью фермента оксидазы гомогетинзиновой кислоты. В норме гомогетинзиновая кислота образуется как промежуточный продукт в процессе обмена фенилаланина и тирозина. В дальнейшем гомо- гетинзиновавя кислота в тканях печени ферментативно расщепляется до фумаровой и ацетоуксусной кислот. При алкаптонурии содержание ГГК в крови и моче повышается в сотни раз. При стоянии такой мочи на воздухе она постепенно окисляется с образованием продукта окисления черного цвета - алкаптона.
Болезнь Хартнупа – наследственная патология, обусловленная нарушением обмена триптофана. Это расстройство касается нарушения процессов всасывания Трп в кишечнике и реабсорбции этой и других нейтральных аминокислот в почках. В результате развиваются: дефицит витамина РР (клиническая картина пеллагры), общая аминаацидурия без увеличения содержания аминокислот в крови, повышение чувствительности к солнечным лучам, двигательная атаксия, характерная для дисфункции мозжечка, усиление образования метаболитов индола в кишечнике под действием микрофлоры. Для лечения используют ниацин как необходимый компонент пищи. Больным рекомендуют защиту от солнечных лучей и периодическую стерили–зацию желудочно-кишечного тракта антибиотиками для подавления образования токсических продуктов метаболизма Трп под действием бактерий.
Пути обмена безазотистого остатка аминокислот: окислительное расщепление аминокислот, глюконеогенез из аминокислот. Гликогенные и кетогенные аминокислоты. Примеры. Биосинтез заменимых аминокислот.
Продуктом дезаминирования аминокислот служит α-кетокислота и аммиак. Большая часть безазотистых остатков аминокислот превращается в пируват непосредственно (Ала, Сер), либо в результате более сложного пути в один из метаболитов ЦТК. Образование аминоациладенилатов. Эта реакция взаимодействия аминокислоты с АТФ, катализируется ферментами класса лигаз. С аминоациладенилатов аминокислоты передаются на тРНК.Углеродные скелеты аминокислот могут включаться в ЦТК. Пять аминокислот (фен, лиз, лей, трп, тир) считаются кетогенными, поскольку являются предшественниками кетоновых тел. Большинство других аминокислот называют гликогенными. Они служат в организме источником углеводов. Глюконеогенез усиливается при сахарном диабете, при гиперфункции коркового вещества надпочечников, введении глюкокортикоидов. В организме человека возможен синтез заменимых аминокислот, к которым относятся: гли, ала, асн, про, сер, глн, глу, асп. Недостаток в пище любой из этих аминокислот не будет сопровождаться её дефицитом в организме. Основными путями образования заменимых аминокислот являются: 1) трансаминирование α-кетокислот, 2) восстановительное аминирование α-кетокислот, 3) синтез с участием незаменимых аминокислот.
Трансаминирование. Источниками атомов углерода в этих реакциях служат метаболиты гликолиза и цикла Кребса, источниками атомов азота – другие аминокислоты, чаще всего – глутамат. Восстановительное аминирование. Источником атома азота аминогруппы является молекула аммиака, источником углерода - α-кетокислоты, чаще всего - α-кетоглутарат Синтез с участием незаменимых аминокислот. Аргинин из цитруллина, тирозин из фенилаланина, цистеин из серина, гистидин при участии глутамина.

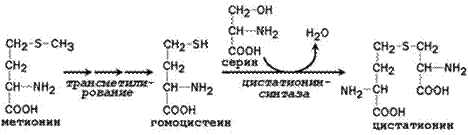
Цистатионин подвергается расщеплению с образованием цистеина и гомосерина, который подвергается дезаминированию в α-кетобутират: