Скорость гомогенной реакции. Кинетическое уравнение реакции. Основные принципы химической кинетики (зависимость скорости от концентрации, суммарная скорость сложной реакции и скорость лимитирующей стадии). Константа скорости. Законы скоростей реакций 1-го и 2-го порядков. Полупериод реакции.
Для химической технологии важен конкретный вид уравнения, позволяющего рассчитывать скорость химической реакции при различных условиях её проведения.
Скорость химической реакции wRJ принято выражать количеством (моль) nJ одного из реагентов или продуктов, прореагировавшем (или образовавшемся) в единицу времени τ в единице реакционного пространства. Под реакционным пространством в гомогенных реакций понимают объём реактора, гетерогенных – поверхность раздела фаз, гетерогенно-каталитических – поверхность раздела фаз или количество катализатора). Для гомогенной химической реакции: W=+- 1dnJ/Vdr, где V - реакционный объём. Если J – исходный реагент, то знак отрицателен, если-продукт, то положителен. Если реакция идёт при постоянном объёме, то скорость можно выразить через молярную концентрацию: CJ=nJ/V. Если в реакции участвует несколько реагентов и получается несколько продуктов, то вводят дополнительно стехиометрический коэффициент j и скорость определяют:
w RJ =± 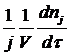 = ±
= ±  , тогда скорость численно будет одинакова по каждому компоненту реакции [w]=моль/м3*с или [w]=моль/л*с.
, тогда скорость численно будет одинакова по каждому компоненту реакции [w]=моль/м3*с или [w]=моль/л*с.
Скорость зависит от большого числа переменных величин: не только от температуры, давления и состава реакционной системы, но и от иных причин, например, посторонних примесей и др.
Факторы, оказывающие влияние на скорость реакций, делятся на две группы: чисто кинетические (микрокинетические), определяющие скорость взаимодействия на молекулярном уровне, и макрокинетические, определяющие влияние на скорость взаимодействия условий транспорта реагентов к зоне реакций – наличие перемешивания, геометрических размеров реактора.
Рассмотрим влияние микрокинетических факторов.
Законы химической кинетики основаны на двух простых принципах (постулатах):
1)скорость химической реакции пропорциональна концентрации реагентов,
2)суммарная скорость нескольких последовательных превращений, разлагающихся по скорости, определяется скоростью наиболее медленной стадии.
Функциональная зависимость скорости химической реакции от концентраций компонентов реакционной смеси w R=f(cA, cB, …cJ) называется кинетическим уравнением реакции. Такое уравнение для элементарной реакции: aA+bB→rR+sS будет: w R=kcAacBb (1), в соответствии с первым постулатом, где k- константа скорости реакции, показатели а и b называются порядками реакций по реагентам А и В. (2) a+b=n – общий порядок. “Молекулярность реакции” равна минимальному числу молекул, одновременно принимающих участие в элементарном акте реакции. Для элементарных реакций порядок равен молекулярности и может иметь значения: 1,2,3. Порядок элементарных реакций не может превышать значения 3, так как вероятность одновременного столкновения более чем трёх молекул чрезвычайно мала. Большинство элементарных реакций – это реакции второго порядка. Используя уравнение (2), можно определить скорость реакций первого и второго порядков, задавая величины a и b. n=1, если a=0,5 и b=0,5; n=2 при a=1, b =1. Простые реакции могут иметь лишь целочисленные значения порядков.
Большинство химических реакций не являются простыми, они протекают через ряд промежуточных стадий. Стехиометрическое уравнение сложной реакции отражает лишь начальное и конечное состояние данной реакционной системы и не описывает механизм реакции. Т. е. сложную реакцию иногда удобно рассматривать как формальную простую и считать, что она протекает в одну, а не в несколько стадий.
Для формально простой реакции: aA+bB→rR+sS, тогда скорость wRA=kcAαcBβ,здесь α и β находят экспериментально и они в общем случае α≠a и β≠b. Полный порядок n=α+β и частные порядки могут быть нецелочленными. Пусть α+β+…=∑nC; если ∑nC=1 – то реакция первого порядка, если ∑nC=2 – то реакция второго порядка, если ∑nC=3 – третьего порядка. Реакции более высоких порядков не наблюдаются.
Наряду с неэлементарными реакциями существует много сложных реакций, которые явно распадаются на стадии (продукты различных стадий образуются в значительных количествах и остаются до конечной стадии). Сложные реакции могут быть параллельными и последовательными. Примером последовательных реакций могут служить реакции расщепления углеводородов с длинной углеводородной цепочкой на всё более мелкие молекулы. Скорость реакции по одному из веществ – её участников – равна алгебраической сумме скоростей тех элементарных стадий, в которых это вещество принимает участие. Скорости стадий компонента J, в которых он расходуется, записываются со знаком плюс, а скорости стадий, в которых компонент J образуется (продукт) записываются со знаком минус, и результирующая скорость будет определяться разностью скоростей на отдельных стадиях, одна из которых будет определять направление реакций и, таким образом, будет являться лимитирующей стадией процесса.
Скорость последовательных стадий в нестационарном режиме различаются между собой, а скорость процесса в целом равна скорости самой медленной стадии, то есть лимитирующей стадии. В стационарном режиме скорость отдельных последовательных стадий “подстраивается” под скорость самой затруднённой стадии; они равны между собой и равны общей скорости процесса: w1= w2=…= wi=…=wz.
Лекция 7.
Интегральные и дифференциальные уравнения законов скоростей реакций. Полупериод реакции. Энергия активации реакции. Влияние температуры на скорость реакции. Способы изменения скорости химической реакции.
Реакции 1-го, 2-го и 3-го порядков описываются кинетическими дифференциальными уравнениями:
-dc/dτ=k’c (1); -dc/dτ=k’’c2 (2); -dc/dτ=k’’’c3 (3), здесь с2=с*с=с1*с2; c3=c*c*c=c1*c2*c3; то есть c1=c2=c3=с. Здесь [c]=моль, с – концентрации веществ 1, 2 и 3.
Преобразуем уравнение (1) dc/с=-k’dτ. После интегрирования получим
ln c=-k’τ+B (4). Постоянную интегрирования найдём, вводя начальные условия: τ=0 и с=со. Тогда уравнение (4) преобразуется: ln co/c= k’τ (5). Введём понятие периода полупревращения (или полупериода реакции – τ1/2), то есть промежутка времени, в течение которого претерпевает превращение половина взятого вещества (τ1/2 - величина, аналогичная периоду полураспада при радиоактивном распаде). Из уравнения (5) получим: τ1/2= ln 2/k’ (6). Здесь мы видим, что период полупревращения не зависит от начальной концентрации реагента (со). Протекание реакции, как правило, не наблюдают до конца, так как для этого требуется длительное время. Практическим окончанием реакции считается время, за которое прореагирует 99,9% исходного вещества. По закону (6) протекают такие процессы, как диффузия и радиоактивный распад (то есть по закону реакций 1-го порядка). Уравнения (5) и (6) называются интегральными уравнениями скорости реакции, так как получены путём интегрирования дифференциального уравнения скорости реакции 1-го порядка. Аналогичным образом получим интегральное уравнение скорости и выражение для периодов полупревращения для реакций 2-го и 3-гопорядков соответственно: τ1/2=  (7) и τ1/2=
(7) и τ1/2=  (8). Используя уравнения (6), (7) и (8) экспериментальным путём определяют порядок реакции.
(8). Используя уравнения (6), (7) и (8) экспериментальным путём определяют порядок реакции.
Проще всего повлиять на соотношение k2/k1 и дифференциальную селективность (при ∆n=0 в формуле (3)) в сторону её увеличения, изменив температуру проведения реакции, так как температура является одним из параметров в наибольшей мере влияющих на скорость химической реакции.
Экспериментально было обнаружено, что при увеличении температуры на 10оС скорость реакции увеличивается в 2-4 раза. Аналитически эта зависимость выражается в виде уравнения Аррениуса: k=koe-E/RT, где k – константа скорости реакции, R – универсальная газовая постоянная, k – предэкспоненциальный множитель, Е – энергия активации.

| Энергия активации (Е) элементарной реакции – это значение кинетической энергии в распределении молекул по энергиям, при которой происходит химическое взаимодействие (энергетический барьер, который должны преодолеть молекулы при переходе из одного состояния реакционной системы в другое).
|
Для обратимой реакции разность энергий активации прямой (Е1) и обратной (Е2) реакций равна тепловому эффекту реакции. Уравнение Аррениуса представляют в виде линейной зависимости логарифма константы скорости от обратной температуры: ln k = f (1/T). Из графика следует, что химические

| реакции более чувствительны к изменению температуры в более низких температурах (∆ln k11) > (∆ln k12) (график 1). Чем выше энергия активации реакции (график 1 наклон >) тем более она чувствительна к температуре.
Если проанализировать селективность для двух параллельных реакций (константы скоростей реакций выразить по уравнению Аррениуса и полученное уравнение продифференцировать по температуре). Из полученного уравнения
|
видно, если энергия активации целевой реакции (Е1) превышает энергию активации побочной реакции, то с ростом температуры наблюдается рост дифференциальной селективности, то есть относительно более быстрое увеличение скорости целевой реакции, то есть скорости процесса. И наоборот, если Е1 < Е2, то для увеличения дифференциальной селективности φ1 нужно понижать температуры.
Из уравнения Аррениуса видно, что возможен ещё один путь управления скоростью реакций – изменение величины Е – энергия активации, если изменить путь реакции, то есть использовать катализаторы.
Как указывалось ранее, скорость химической реакции зависит от большого числа факторов: прежде всего, скорость простой реакции пропорциональна концентрации веществ-реагентов. На изменение скорости реакции в зависимости от концентрации существенно влияет порядок реакции. Для сложных реакций, в частности для параллельных, при которых получаются целевой и побочные продукты, анализ соотношения скоростей проводят, используя дифференциальную селективность (равную отношению скорости расходования реагента А на целевую реакцию к общей скорости расходования реагента и на целевую, и на побочные реакции). Если φ1(СА) – возрастающая функция, то скорость целевой реакции с ростом концентрации исходного реагента возрастает быстрее скорости побочного продукта (при n1 > n2). И, наоборот, при (n1 < n2), φ1(СА) – убывающая функция – более высокая дифференциальная селективность по целевому продукту достигается при низкой концентрации исходного реагента. При n1=n2, φ1(СА) – остаётся постоянной в зависимости от концентрации.
Лекция 8.