Хромосомные мутации характеризуются нарушением структуры хромосом.
Механизм возникновения: 1) разрыв хромосомы 2) неравномерный кроссинговер

Причины возникновения: 1) разрывы хромосом и хроматид и воссоединение в новых сочетаниях 2) резкие отклонения в нормальном течении клеточного цикла
Хромосомные болезни - это большая группа врожденных наследственных болезней, клинически характеризующихся множественными врожденными пороками развития. В их основе лежат хромосомные или геномные мутации.
Синдром Дауна- геномная мутация(трисомия по 21 паре хромосом)
Синдром Патау – геномная мутация (трисомия по 13 хромосоме)
Синдром Эдвардса- геномная мутация(трисомия по 18 паре хромосом)
Синдром трисомия по Х- геномная мутация
Синдром Клайнфельтера – геномная мутация (трисомия или полисомия по 23-й паре хромосом)
Синдром Шершевского – Тернера – геномная мутация(моносомия)
Синдром «кошачьего крика» -хромосомная мутация(делеция)
Транслокционный синдром Дауна- хромосомная мутация(транслокация)
61. Генные мутации, причины, классификация.
62. Мутации по типу ошибки репликации, механизмы развития.
63. Мутации по типу сдвига рамки считывания, механизмы развития.
64. Механизмы развития наследственных болезней при генных мутациях.
65. Репарация, причины, типы.
66. Механизмы возникновения репарации.
67. Апоптоз, определение.
68. Причины апоптоза.
69. Стадии развития апоптоза.
70. Генетический контроль процесса апоптоза.
Нарисовать и объяснить причины и механизмы возникновения генных(точковых) мутаций
Точечная мутация — тип мутации в ДНК или РНК, для которого характерна замена одного азотистого основания другим. Термин также применяется и в отношении парных замен, инсерции или делеции одного или нескольких нуклеотидов. Точечные мутации, возникающие в некодирующей ДНК, обычно никак себя не проявляют. Точечный мутант — организм, в генотипе которого произошла точечная мутация.
Точечные мутации классифицируют по эффекту, который изменённый нуклеотид оказывает на триплет:
· Сеймсенс-мутация — кодон продолжает кодировать ту же аминокислоту.
· Нонсенс-мутация — мутация, в результате которой кодон теряет способность кодировать какую-либо аминокислоту и становится стоп-кодоном, что приводит к преждевременной терминации синтеза белка.
· Миссенс-мутация — переключает кодон на кодирование другой аминокислот
Причины возникновения Точечные мутации могут возникать в результате спонтанных мутаций, происходящих во время репликации ДНК. Они также могут возникать в результате действия мутагенов — к примеру, воздействия ультрафиолетового или рентгеновского излучения, высокой температуры или химических веществ. Мутации появляются при синтезе молекулы ДНК, содержащей повреждения, в процессах репликации ДНК, репарации ДНК или транскрипции
Нарисовать и объяснить причины и механизмы возникновения аутосомных и гоносомных синдромов человека
Аутосомный синром
Синдром Дауна. Первое клиническое описание синдрома Дауна относится к 1866г. Частота 1: 550 - 650 новорожденных. Среди умственно отсталых детей выявляется 10-12% больных с синдромом Дауна.
При цитогенетическом анализе выявляют 3 формы:
- простая трисомия по хромосоме 21 (в 95% случаев)
- транслокация хромосомы 21 на другие хромосомы (чаще на 14, 15, реже на 21, 22 и Y-хромосому) (4% случаев)
- мозаичный вариант синдрома (1 %).
Больные с-м Дауна имеют характерный фенотип: брахицефалическая форма черепа с укорочением передне-заднего размера и утолщением затылка; раскрытые роднички, избыток кожи на затылке, плоский профиль лица, характерный разрез глаз, помутнение хрусталика, нистагм, короткий нос с широким переносьем, полуоткрытый рот с толстыми губами и большим языком, Руки короткие и широкие, клинодактилия мизинцев, часто наблюдаются врожденные пороки сердца, деформации скелета, задержка психомоторного развития, с возрастом нарастает интеллектуальный дефицит.
При транслокации симптоматика более выражена, чем при обычной трисомии. В кариотипе обнаруживают 46 хромосом, одну обычную хромосому 21 и одну маркерную хромосому, включающую хромосому 21, транслоцированную на другую хромосому.
Анализ кариотипов родителей детей с транслокационной формой синдрома нередко выявляет у фенотипически нормальной матери пробанда 45 неизмененных хромосом и наличие маркерной хромосомы - точно такой же, как у её больного ребенка.
При мозаичной форме синдрома выраженность фенотипа зависит от доли трисомных клеток в аномальном кариотипе пробанда: чем их больше, тем более выражен фенотип синдрома.
Синдром Патау. Эта трисомия по хромосоме 13 встречается с частотой 1: 6000 новорожденных. Цитогенетически различают 3 формы: трисомная (75%), транслокационная (20%) и мозаичная (5%). Фенотип включает в себя прежде всего, триаду признаков: микрофтальм, расщелины губы и неба, полидактилия. Черепно-лицевой дисморфизм характеризуется также микроцефалией, черепом неправильной формы, скошенным узким лбом, раскрытыми родничками и швами черепа, дефектами скальпа, узкими горизонтальными щелями. Нос плоский,широкий, переносье запавшее. Ушные раковины расположены низко.
При с-ме Патау центральная нервная система поражена во всех случаях. Характерно множественное поражение внутренних органов: пороки сердца, поликистоз почек, аномалии желудочно-кишечного тракта. Продолжительность жизни больных резко снижена. Обычно дети погибают в первые дни или недели жизни.
Синдром «кошачьего крика». Синдром делеции часто короткого плеча хромосомы 5. Частота синдрома - 1: 650 000; среди детей с задержкой умственного развития - 1: 350. Описаны мозаичные формы.
Дети с этим синдромом, как правило, рождаются после нормально протекавшей беременности. В неонатальном периоде состояние резко ухудшается: приступы посинения, удлиненный выдох, снижение двигательной активности, угнетение сосательного рефлекса, рвота.
Фенотип характеризуется монотонным или резким криком, похожим на кошачье мяуканье. С возрастом крик исчезает. Выявляется грубое физическое и интеллектуальное недоразвитие. Выражен черепно-лицевой дисморфизм (особенно в первые 2 года жизни): микроцефалия, вытянутая форма черепа, лунообразное лицо, узкие глазные щели, вдавленное переносье. Зубы расположены неправильно, передние резцы выступают вперед, рот широкий, толстая нижняя губа. Часто наблюдаются врожденные пороки сердца, почек, косолапость, сращение 2 и 3-го пальцев ног и некоторые другие признаки
45. Характеристика гоносомных синдромов Гоносомные синдромы подразделяются на синдромы с мужским и женским фенотипами, а также на клинические формы гермафродитизма.
Полисомия по У - хромосоме. Это хромосомный синдром с мужским фенотипом, общее название - с-м Клайнфельтера. Классическая форма - 47, ХХУ, составляющая 80% всех случаев синдрома, частота в популяции 1: 500, на оставшиеся 20% приходятся 48 ХХХУ и варианты мозаичной формы синдрома. Общим для всех вариантов и форм является триада признаков: гинекомастия, атрофия и гипоплазия тестикул и бесплодие. Вследствие гормонального дисбаланса такие больные имеют высокий рост, узкие плечи, широкий таз, скудное оволосение. Уровень гонадотропинов в моче соответствует норме или повышен.
Синдром полной моносомии по Х-хромосоме. Это синдром с женским фенотипом. Единственным примером этого синдрома является с-м Шерешевского - Тернера. Частота синдрома 1: 1430 у новорожденных девочек. Классический вариант - 45, ХО (55% всех случаев). Возможны варианты:
- мозаики 45, ХО/46, ХХ; 45, ХО/47, ХХХ
- делеции: 45, ХО, del (Х)
- кольцевая хромосома: 45, ХО, r (Х)
- другие варианты.
Фенотипически это лица женского пола с задержкой роста и полового развития. У них нормальные наружные и недоразвитые внутренние половые органы. Характерный признак - короткая шея с низкой линией роста волос на затылке и крыловидными складками кожи на шее.
30) Нарисрвать и объяснить причины и механизмы возникновения моносомий и трисомий у человека Синдром полной моносомии по Х-хромосоме. Это синдром с женским фенотипом. Единственным примером этого синдрома является с-м Шерешевского - Тернера. Частота синдрома 1: 1430 у новорожденных девочек. Классический вариант - 45, ХО (55% всех случаев). Возможны варианты:
- мозаики 45, ХО/46, ХХ; 45, ХО/47, ХХХ
- делеции: 45, ХО, del (Х)
- кольцевая хромосома: 45, ХО, r (Х)
- другие варианты.
Фенотипически это лица женского пола с задержкой роста и полового развития. У них нормальные наружные и недоразвитые внутренние половые органы. Характерный признак - короткая шея с низкой линией роста волос на затылке и крыловидными складками кожи на шее.
Трисомия
Трисомия — это наличие трёх гомологичных хромосом вместо пары (в норме). Причиной подавляющего большинства трисомий у человека являются ошибки расхождения хромосом при оогенезе, причём наибольший вклад дают ошибки в мейозе I по сравнению со вторым мейотическим делением. Вероятность трисомий у потомства повышается с возрастом матери. Единственной жизнеспособной трисомией по аутосоме у человека является трисомия по хромосоме 21, вызывающая синдром Дауна. Трисомики по хромосомам 13 (синдром Патау) и 18 (синдром Эдвардса) могут дожить до рождения
35) Основным свойством раковой клетки является ее безудержное и неконтролируемое деление, обусловленное отсутствием тормозящих пролиферацию клеток механизмов и факторов. Злокачественные опухоли являются по происхождению моноклональными, т е все клетки опухоли происходят от одной-единственной клетки – предшественницы и являются ее потомками. Автономность. Все злокачественные опухоли растут "сами по себе", они не подчиняются, не реагируют на различные (нервные, гуморальные) сигналы организма-хозяина. Клетки такой опухоли практически полностью утрачивают зависимость от внешних и внутренних факторов, они представлены сами себе. Склонность к проникновению («инвазии», «инфильтрации», «пенетрации») в окружающие ткани, с формированием местных метастазов.
Склонность к метастазированию в другие, часто весьма отдалённые от исходной опухоли ткани и органы посредством перемещения по лимфо- и кровеносным сосудам, а также имплантационно. Причём определённые типы опухолей проявляют определённое родство («тропность») к определённым тканям и органам — метастазируют в определённые места (но могут метастазировать и в другие).
34) Существует определенный класс генов, вызывающих злокачественную трансформацию клеток. Эти гены называются онкогенами. Протоонкогенами называются нормальные аналоги или предшественники окогенов.Превращение протоокогенов в онкогены осущ путем:
1) присоедеинения к протоокогену нового транскрипционного промотора
2) амплификация(увеличения числа копий) протоонкогена до онкогенной активности
3) включения в молекулу ДНК клетки хозяина некоторой последовательности нуклеотидов, усиливающей действие промотора
4) присоединение протоонкогена к локусу иммуноглобулина в результате транслокации
5) мутации протоонкогена
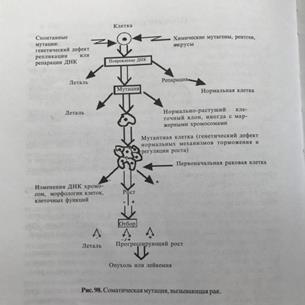
33) Живые организмы и их клетки не вечны: рано или поздно их существование заканчивается гибелью. Это естественный, генетический запрограммированный процесс, получивший название апоптоза. В общем виде процесс апоптоза можно представить следующим образом
Нарушение структуры и функционирования клеток – апоптоз - гибель клеток – элиминация.
В процессе апоптоза принимают участия специфические ферменты каспазы (бывают сериновые и цистеиновые) В нормальных клетках они представлены неактивными предшественниками прокаспазами. Они активируется в случае нарушение структуры клеток.
Апоптоз по команде: вызывается внешней сигнализацией, которая передается через мембранные рецепторы. Вызывается если отсутствует один из факторов:1) ростовой фактор 2)прикрепление 3) контактное торможение. Первым в ответ на сигнал активируется К8. Сигнал о нарушениях структур клетки может поступать также из митохондрий. Сигнальным фактором в этом случае является протеаза AIF. Он способствует превращению неактивной формы прокаспазы 9 в активную каспазу 9. В процессе активизации прокаспазы 9 принимает участие Цитохром С, стимулирующий взаимодействие прокаспазы 9 с белком Apaf 1 т другими прокаспазами 9. Ключевым ферментом апоптоза является каспаза 3, активируемая каспазами 8 и 9. Каспаза 3 активирует другие каспазы 6 и 7, и некоторые другие некаспазные белки.
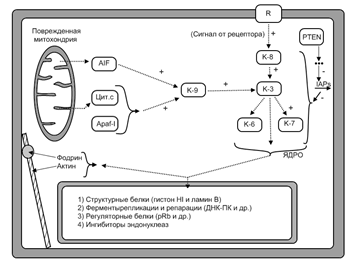
Апоптоз изнутри Может происходит из за нездорового состояния клетки, чрезмерные повреждений хромосом, повреждение внутриклеточной мембраны. Белок р53 является одним из ключевых белков апоптоза. В нормальной клетке количество и активность белка р53 поддерживается на минимальном уровне. Концентрация и активность белка р53 контролируется белком Mdm2, который при взаимодействии с белком с белком р53 снижает его активность и ускоряет его распад. Избыток белка р53 приводит к активации белка Mdm2 с последующим снижением активности и ускорением распада белка р53. Регуляторный фактор белок Arf или 19Arf задерживает распад белка р53. Еще один регуляторный белок 14-3-3в служит активатором белка р 53. Высокая активность белка р 53 в то время как белок Mdm2 регулирует как концетрацию так и активность белка р53.

32). Пострепликативная репарация Наименее изученный тип репарации. Пострепликативная репарация осуществляется путем рекомбинации (обмена фрагментами) между двумя вновь образованными двойными спиралями ДНК. Примером такой пострепликативной репарации может служить восстановление нормальной структуры ДНК при возникновении тиминовыхдимеров (Т-Т), когда они не устраняются самопроизвольно под действием видимого света (световая репарация) или в ходе дорепликативнойэксцизионной репарации.
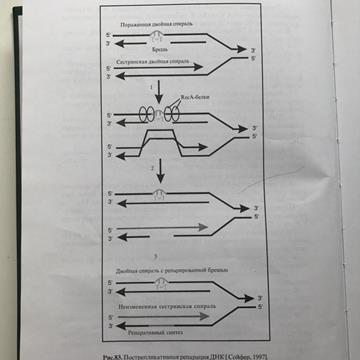
Важным механизмом удаления повреждений в ДНК является эксцизионная репарация (1).Специфическая нуклеаза удаляет небольшой сегмент ДНК, включающий поврежденный участок. Удаленный участок восстанавливается ДНК-полимеразой, использующей в качестве матрицы комплементарную цепь. Наконец, оставшийся одноцепочечный разрыв закрывается ДНК-лигазой.Тиминовыедимеры могут быть удалены фотореактивацией (2). Специфическая фотолиаза связывается с дефектным участком ДНК и после облучения расщепляет димер с образованием отдельных нуклеиновых оснований. Третий механизм — это репарация в результате рекомбинации (3, показано в упрощенном виде). В этом процессе участок, содержащий повреждение, пропускается во время репликации. Образующаяся брешь закрывается путем сдвига соответствующего сегмента из правильно реплицированной второй цепи. Новая брешь ликвидируется с участием полимераз и ДНК-лигаз. В завершение первоначальный дефект (1) устраняется путем вырезания.
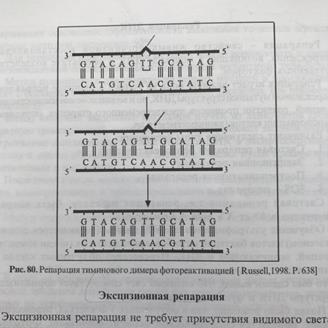
31) 1.Световая репарация. Механизм это репарации заключается в том, что под действием ультрафиолетовых лучей, в молекуле ДНК могут возникать сцепления между соседними пиримидиновыми основаниями с образованием димеров – Т=Т, Т=Ц, Ц=Ц, Ц=У, Т=У, У=У. квант видимого света активизирует специальный фермент, которые соединяются с поврежденной ДНК, разъединяют димеры и восстанавливает целостность нити ДНК.
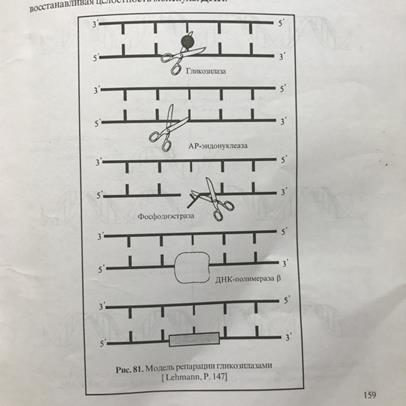
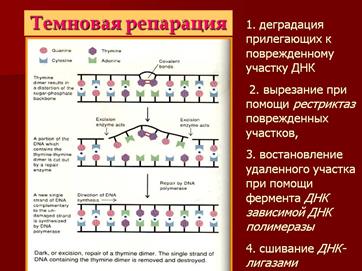
2.Темновая репарация. Клетки могут ликвидировать повреждения ДНК и без участия света. Стадии: а)узнавание повреждения ДНК, при помощи фермента нуклеазы б)вырезания, с помощью экзонуклеазы в) матричный синтез новой цепи с помощью полимераз г) соединение новообразованного участка.
19. Клинико-генеалогический метод включает клиническое обследование членов семьи пациента, обратившегося за консультацией, составление ее родословной и проведение генеалогического анализа. Генеалогический анализ является самым распространенным, наиболее простым и одновременно высоко информативным методом, доступным каждому, кто интересуется своей родословной и историей своей семьи. Он не требует никаких материальных затрат и аппаратуры. Убеждены, что со временем в каждой истории болезни будет представлена родословная пациента, как обязательная часть анамнеза жизни.
20.
21. Методы пренатальной диагностики могут быть разделены на 2 группы:
· косвенные – когда об имеющейся у плода патологии судят по изменениям в организме матери;
· прямые – когда объектом исследования является сам плод.
К косвенным методам пренатальной диагностики относятся способы оценки фетоплацентарной системы путем определения в крови матери ряда гормонов (эстрогенов, прогестерона, хорионического гонадотропина), некоторых специфических белков (плацентарного лактогена, трофобластического бета-1‑гликопротеина, плацентарного протеина‑5 и др.) и ферментов (плацентарной щелочной фосфатазы, окситоциназы). С помощью этих методов диагностируется фетоплацентарная недостаточность, что обеспечивает возможность проводить коррекцию выявленных нарушений, тем самым осуществляя профилактику внутриутробной гипоксии, гипотрофии и антенатальной гибели плода.
В настоящее время с целью скринирующего обследования всех беременных женщин применяется двукратное исследование в сыворотке крови фетального белка – альфа–фетопротеина (АФП) – на 14–16 и 21–22 неделях гестации. Значительное увеличение АФП происходит при анэнцефалии, spina bifida, черепно-мозговых грыжах, атрезиях желудочно-кишечного тракта, врожденных заболеваниях почек, многоплодной беременности, в случае гибели плода. Снижение уровня АФП возможно при хромосомной патологии плода, плацентарной недостаточности, внутриутробной гипотрофии.
В настоящее время для диагностики патологических состояний у плода проводится трехмаркерный скрининг беременных женщин (наряду с АФП определяется уровень β‑хорионического гонадотропина и эстриола). β‑хорионический гонадотропин (β‑субъединица хориогенина человека) повышается при многоплодной беременности, резус–конфликте, хромосомных синдромах у плода. Низкие значения отмечены при хронической плацентарной недостаточности, замершей беременности, антенатальной гибели плода. Снижение уровня гормона может быть связано с угрожающим выкидышем и внематочной беременностью.
Уровень эстриола в сыворотке крови женщины нарастает в соответствии со сроком беременности и степенью увеличения размера плода. Высокое содержание гормона отмечается при крупном плоде и многоплодной беременности. Низкое содержание эстриола отмечается при фетоплацентарной недостаточности, внутриутробной гибели, гипотрофии плода, врожденных пороках сердца, синдроме Дауна у плода.
При необходимости ряду беременных женщин проводятся дополнительные исследования, в том числе определение в сыворотке крови плацентарного лактогена (ПЛ). Увеличение концентрации ПЛ может наблюдаться при резус–конфликтной беременности, многоплодии, крупном плоде.
22. Медико-генетическое консультирование является наиболее распространенным видом профилактики наследственных болезней. Суть его заключается в прогнозировании рождения ребёнка с наследственной патологией, объяснении вероятности этого события консультирующимся и помощи семье в принятии решения о дальнейшем деторождении. Медико-генетическое консультирование как способ профилактики врождённой или наследственной патологии особенно эффективен до зачатия или на самых ранних сроках беременности[1] :1.
Включает 3 этапа:
1. Уточнение диагноза с использованием специальных генетических методов: генеалогическое обследование и составление родословной, биохимико-генетические методы, позволяющие выявить генетически обусловленные изменения обмена веществ, диагностика гетерозиготного носительства рецессивных аллелей, пренатальная диагностика (УЗИ, биохимический скрининг маркерных белков в сыворотке беременной, амниоцентез — забор околоплодной жидкости для кариотипирования плода).
2. Определение прогноза потомства, который основывается на данных о типе и варианте наследования патологического состояния, результата пренатальной диагностики.
3. Формулирование заключения и объяснение заинтересованным лицам в доступной форме смысла генетического риска[2].
В идеальном варианте медико-генетическое консультирование должны пройти все семьи, планирующие иметь ребёнка (т.н. проспективное консультирование). Прямыми показаниями для направления к специалисту-генетику являются:
· установленная или подозреваемая наследственная болезнь в семье;
· кровнородственные браки;
· воздействие возможных мутагенов или тератогенов до или в течение первых трёх месяцев беременности;
· значимые отклонения результатов биохимического скрининга маркерных сывороточных белков у беременной;
· выявление у плода маркёров хромосомных болезней и врождённых пороков развития при ультразвуковом исследовании[1]
23. Генетический риск — это вероятность появления определенной наследственной патологии у обратившегося за консультацией или у его потомков. Он определяется путем расчетов, основанных на анализе генетических закономерностей, или с помощью эмпирических данных. Возможность рассчитать генетический риск зависит в основном от точности диагноза и полноты генеалогических данных.
Генетический риск до 5 % оценивается как низкий и не считается проти-вопоказанием к деторождению в данной семье. Риск от 6 до 20 % принято считать средним. В этом случае рекомендации относительно планирования дальнейших беременностей зависят от тяжести медицинских и социальных последствий конкретного наследственного заболевания и от возможности проведения пренатальной диагностики. Риск более 20 % считается высоким, и в отсутствие методов пренатальной диагностики соответствующей патологии дальнейшее деторождение в данной семье не рекомендуется.
Охарактеризовать множественные аллели на примере групп крови человека по сиситеме АВ0: гены, генотипы, фенотипы
Развитие признака определяется двумя аллелями одного гена (А и а), которые занимают идентичные локусы гомологичных хромосом. Иногда ген имеет не два, а большее число аллелей, которые возникают в результате мутации. Многократные мутации одного и того же гена образует серию множественных аллелей, а само явление называется явлением множественного аллелизма.
Таким образом, множественный аллелизм — это существование в популяции более двух аллелей данного гена. Оно имеет широкое распространение: окраска шерсти у кроликов, собак, цвет глаз у дрозофилы, система групп крови АВО у человека.
Имеются определенные закономерности множественного аллелизма:
— каждый ген может иметь большое число аллелей;
— любой аллель может возникнуть в результате прямой и обратной мутации любого члена серии множественных аллелей или от аллеля дикого типа;
— в диплоидном организме могут одновременно находиться два любых аллеля из серии множественных аллелей;
— аллели находятся в сложных доминантно-рецессивных отношениях между собой: один и тот же аллель может быть доминантным по отношению к одному аллелю и рецессивным по отношению к другому, а между иными аллелями доминирование может отсутствовать, и наблюдается кодоминирование и др.;
— члены серии множественных аллелей наследуются так же, как и пара аллелей, т. е. наследование подчиняется менделевским закономерностям (кроме кодоминирования);
— разные сочетания аллелей в генотипе обуславливают различные фенотипические проявления одного и того же признака;
— серии аллелей увеличивают комбинатов ну ю изменчивость.
Примером множественного аллелизма у человека является наличие трех аллелей гена, определяющего наследование групп крови системы АВО.
- система определяется тремя аллелями одного гена I (IA, 1В, 10); ген I расположен в 9-й хромосоме: 9q34;
- из всей серии аллелей одновременно в генотипе диплоидного организма находятся только две аллели, делая возможным следующие генотипы: I0I0; IAIA; IAI0; IВIВ; IВI0; IAIВ;
- аллели IA, IВ доминантны по отношению к аллелю I0 по принципу полного доминирования, в свою очередь, между собой аллели IА и IВ — кодоминантны;
- различные сочетания аллелей в генотипе дают разные фенотипы: 4 группы крови I (0), II (А), III (В), IV (АВ), которые различаются между собой антигенными свойствами эритроцитов. Антигены (агглютиногены) находятся на поверхности эритроцитов (гликокаликс);
- доминантный аллель гена может проявлять свое действие в гомо- (IAIA, IBIB – II и III группы соответственно) и гетерозиготном состояние (IАI0, IВ I0 – II и III группы соответственно), а рецессивный аллель гена — только в гомозиготном (I0I0 – I группа), при кодоминирование проявляется новый фенотипический признак (IAIВ – IV группа);
- особенностью системы является наличие в сыворотке крови специфических антител (агглютининов), разноименных по отношению к собственным агглютиногенам (они одновременно находятся в крови);
Дать определение пенентрантности, экспрессивности, плейотропии. Привести примеры.
В популяционной генетике пенетрантность — это показатель фенотипического проявления аллели в популяции. Определяется как отношение (обычно — в процентах) числа особей, у которых наблюдаются фенотипические проявления наличия аллели, к общему числу особей, у которых данная аллель присутствует в необходимом для фенотипического проявления количестве копий (в зависимости от характера доминирования, для фенотипического проявления может быть достаточно только одной копии аллеля или двух, если для фенотипического проявления необходимо, чтобы особь была гомозиготна по данному гену).
Если частота экспрессии фенотипа менее 100%, т.е. существуют лица, имеющие соответствующий генотип без каких-либо его проявлений, говорят, что ген имеет неполную пенетрантность. Полная пенетрантность — это 100 % фенотипическое проявление наличия данного аллеля в пределах популяции. Проще говоря, пенентрантность - это частота проявления гена в признаках.
К примеру, ген I – отвечающий за наследование группы крови обладает полной пенетрантностью. В качестве примера неполной пенетрантности иногда приводят полидактилию
Экспрессивность — тяжесть экспрессии фенотипа среди индивидуумов с одним патологическим генотипом. Когда тяжесть болезни различается у людей, имеющих тот же генотип, говорят, что фенотип имеет вариабельную экспрессивность. Даже в одной родословной два индивидуума, несущих те же мутантные гены, могут иметь некоторые одинаковые признаки и симптомы, а другие проявления болезни могут различаться в зависимости от пораженных тканей и органов.
Количественные показатели экспрессивности измеряются на основе статистических данных.Например, на всех растениях пшеницы, гомозиготных по гену, обусловливающему отсутствие остей, развиваются безостые колосья. Другие же гены (и их, по-видимому, большинство) отличаются изменяющейся Э. У кроликов и некоторых других животных известен рецессивный ген гималайской («горностаевой») окраски, обусловливающей своеобразную пятнистость меха (на белом или светлом фоне кончики лап, ушей, морды и хвоста имеют чёрную окраску). Однако такая окраска развивается только при выращивании молодняка гималайской породы при умеренных температурах. При повышенной температуре весь мех у особей того же гималайского генотипа оказывается белым, а при пониженной — чёрным. Этот пример указывает на то, что на Э. влияют факторы внешней среды, в данном случае температуры. При одинаковых условиях внешней среды Э. гена может варьировать в зависимости от генотипической среды, т. е. от того, в сочетании с какими другими генами данный ген входит в состав генотипа. Плейотропи́я — явление множественного действия гена. Выражается в способности одного гена влиять на несколько фенотипических признаков. Таким образом, новая мутация в гене может оказать влияние на некоторые или все связанные с этим геном признаки. Этот эффект может вызвать проблемы при селективном отборе, когда при отборе по одному из признаков лидирует один из аллелей гена, а при отборе по другим признакам — другой аллель этого же гена. Выделяют первичную (ген одновременно проявляет множественное действие - синдром Марфана обусловлен действием одного гена) и вторичную (имеется одно первичное фенотипическое проявление гена, которое обуславливает проявление вторичных признаков. Например, аномальный гемоглобин S в гомозиготном состоянии фенотипически первично проявляется в виде серповидноклеточной анемии, которая приводит к вторичным фенотипическим проявлениям в виде невосприимчивости к малярии, анемии, гепатолиенальному синдрому, поражению сердца и мозга)
Дать определение, нарисовать и объяснить сущность и медицинское значение полярности яйцеклетки, ооплазматической сегрегации, позиционной информации, детерминации и дифференциации в раннем онтогенетическом развитии.
Полярность яйцеклеток намечается еще на стадии накопления желтка в овоцитах во время их быстрого (большого) роста и закрепляется при выделении полярных (редукционных) телец. После выделения второго редукционного тельца полярность становится устойчивой и необратимой, что доказывается опытами Геррье по центрифугированию яйцеклеток на разных стадиях их созревания. Полюс, на котором выделяются редукционные тельца, называется анимальным (буква А на схеме), а противоположный ему - вегетативный. Полюса яйцеклетки отличаются по многим параметрам: концентрации различных веществ, количеству органоидов, активности протекания внутриклеточных процессов и ряду других. 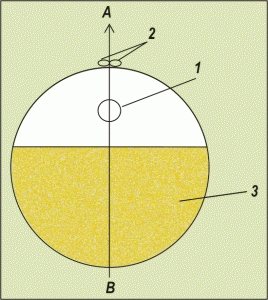 Анимальный полюс – это точка поверхности яйцеклетки, где от нее отделяются редукционные тельца. Как известно, яйцеклетка животных образуется в результате мейоза. Мейоз предшественника яйцеклетки (ооцита) неравномерен: там получается одна яйцеклетка и три совсем маленькие клетки, которые в подавляющем большинстве случаев в дальнейшем развитии не участвуют. Эти маленькие клетки традиционно называют редукционными тельцами (они же полярные, они же направительные тельца). На картинке они обозначены цифрой 2. Вблизи анимального полюса яйцеклетки находится ядро (цифра 1). Точка яйцеклетки, противоположная анимальному полюсу, называется вегетативным полюсом (буква В). В области вегетативного полюса скапливаются запасные вещества, нужные для питания зародыша, т.е. желток (цифра 3). На анимальном полюсе желтка меньше всего, там – ядро в окружении чистой цитоплазмы. Анимальный полюс более физиологически активен, именно с него начинается деление яйцеклетки. Там, где желтка очень много, его объем может во много раз превосходить объем анимальной чистой цитоплазмы (пример – птичье яйцо). В некоторых яйцеклетках исчезает даже и анимально-вегетативная полярность (пример – яйцо насекомых).
Анимальный полюс – это точка поверхности яйцеклетки, где от нее отделяются редукционные тельца. Как известно, яйцеклетка животных образуется в результате мейоза. Мейоз предшественника яйцеклетки (ооцита) неравномерен: там получается одна яйцеклетка и три совсем маленькие клетки, которые в подавляющем большинстве случаев в дальнейшем развитии не участвуют. Эти маленькие клетки традиционно называют редукционными тельцами (они же полярные, они же направительные тельца). На картинке они обозначены цифрой 2. Вблизи анимального полюса яйцеклетки находится ядро (цифра 1). Точка яйцеклетки, противоположная анимальному полюсу, называется вегетативным полюсом (буква В). В области вегетативного полюса скапливаются запасные вещества, нужные для питания зародыша, т.е. желток (цифра 3). На анимальном полюсе желтка меньше всего, там – ядро в окружении чистой цитоплазмы. Анимальный полюс более физиологически активен, именно с него начинается деление яйцеклетки. Там, где желтка очень много, его объем может во много раз превосходить объем анимальной чистой цитоплазмы (пример – птичье яйцо). В некоторых яйцеклетках исчезает даже и анимально-вегетативная полярность (пример – яйцо насекомых).
Ооплазматическая сегрегация: перераспределение биологически активных молекул (локальных детерминант) в цитоплазме яйцеклетки в результате ее активации.
При этом разнородные по составу участки цитоплазмы попадают в клетки, которые дают начало разным зачаткам. Так, наиболее богатые желтком вегетативные участки яйца попадают при дроблении в бластомеры, которые дают начало энтодерме.
Особое значение для судьбы развивающегося зародыша имеет, однако, сегрегация инструктивных молекул, наблюдаемая в период дробления у многих животных. Материнские цитоплазматические факторы, попадая в те или иные бластомеры, активируют специфические программы развития и детерминируют разнообразные зачатки.
Показано, что поведение клетки меняется в зависимости от ее расположения в составе зародыша. Это означает, что клетки способны воспринимать и запоминать позиционную информацию. Позиционная информация - это какие-то сигналы, которые сообщают клетке о ее положении в зародыше. Чаще всего, видимо, такими сигналами служит концентрация биологически активных веществ - морфогенов.
В зависимости от полученных сигналов клетка реализует ту или иную программу развития. Однако выбор программы происходит не один- единственный раз: по ходу развития клетка много раз становится перед проблемой выбора пути. Дело в том, что сигналы, которые могут служить позиционной информацией, распространяются обычно на очень небольшие расстояния порядка сотен или тысяч клеточных диаметров, т.е. около 1-2 мм. Поэтому, пока зародыш имеет длину около 1 мм, у него закладываются передний и задний конец, спинная и брюшная стороны: клетки "узнают", к какой части тела (например, передней или задней) они относятся. Когда зародыш подрастет, могут поступать инструкции о более мелких деталях: например, клетки определенной области узнают, что они "должны" стать клетками конечности. Затем, в зависимости от положения внутри зачатка конечности, клетки получают сигналы о том, должны ли они стать клетками скелета или мышц, плеча или запястья и т. п.
Дифференциация — это стойкое структурно-функциональное преобразование клеток в различные специализированные клетки. Дифференцировка клеток биохимически связана с синтезом специфических белков, а цитологически — с образованием специальных органелл и включений. При дифференцировке клеток происходит избирательная активация генов. Важным показателем клеточной дифференцировки является сдвиг ядерно-цитоплазменного отношения в сторону преобладания размеров цитоплазмы над размером ядра. Дифференцировка происходит на всех этапах онтогенеза. Особенно отчетливо выражены процессы дифференциации клеток на этапе развития тканей из материала эмбриональных зачатков. Специализация клеток обусловлена их детерминацией. Детерминация — это процесс определения пути, направления, программы развития материала эмбриональных зачатков с образованием специализированных тканей. Детерминация может быть оотипической (программирующей развитие из яйцеклетки и зиготы организма в целом), зачатковой (программирующей развитие органов или систем, возникающих из эмбриональных зачатков), тканевой (программирующей развитие данной специализированной ткани) и клеточной (программирующей дифференцировку конкретных клеток). Различают детерминацию: 1) лабильную, неустойчивую, обратимую и 2) стабильную, устойчивую и необратимую. При детерминации тканевых клеток происходит стойкое закрепление их свойств, вследствие чего ткани теряют способность к взаимному превращению (метаплазии). Механизм детерминации связан со стойкими изменениями процессов репрессии (блокирования) и экспрессии (деблокирования) различных генов.