

Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...

Архитектура электронного правительства: Единая архитектура – это методологический подход при создании системы управления государства, который строится...
Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Архитектура электронного правительства: Единая архитектура – это методологический подход при создании системы управления государства, который строится...
Топ:
Характеристика АТП и сварочно-жестяницкого участка: Транспорт в настоящее время является одной из важнейших отраслей народного хозяйства...
Особенности труда и отдыха в условиях низких температур: К работам при низких температурах на открытом воздухе и в не отапливаемых помещениях допускаются лица не моложе 18 лет, прошедшие...
Организация стока поверхностных вод: Наибольшее количество влаги на земном шаре испаряется с поверхности морей и океанов...
Интересное:
Как мы говорим и как мы слушаем: общение можно сравнить с огромным зонтиком, под которым скрыто все...
Национальное богатство страны и его составляющие: для оценки элементов национального богатства используются...
Наиболее распространенные виды рака: Раковая опухоль — это самостоятельное новообразование, которое может возникнуть и от повышенного давления...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Для того чтобы получить абсолютный спирт, необходимо провести обезвоживание 96% спирта при помощи прокаленного медного купороса (сульфат меди). В основе этого метода лежит свойство сульфата меди отдавать и присоединять молекулы воды, меняя при этом свой цвет (прокаленный сульфат меди имеет вид серовато-белого порошка; который по мере присоединения воды приобретает синюю окраску). Насыпав порошок прокаленного медного купороса (примерно 10 г на 100 мл спирта) в чистую стеклянную бутылку с притертой пробкой, наливают туда же 96% спирт. Затем бутылку встряхивают до равномерного распределения порошка. Подобную процедуру повторяют на протяжении 1—2 дней. По мере поглощения воды порошок приобретает синюю окраску. Однократная обработка, как правило, не дает обезвоживания, поэтому спирт переливают в другой сосуд, содержащий свежую порцию сульфата меди. Подобную процедуру повторяют до тех пор, пока осадок не перестанет приобретать голубой цвет. Обезвоженный спирт отфильтровывают в чистую посуду, которую плотно закрывают. Желательно проверить рН абсолютного спирта, как после обработки сульфатом меди он может стать слегка подкисленным. Для нейтрализации достаточно прибавить небольшое количество карбоната кальция (СаСОз). Если в лаборатории нет фабричного порошка безводного сульфата меди, то его можно приготовить самим, прокалив кристаллическую соль над огнем в широкой фарфоровой чашке (до тех пор, пока из синих кристаллов не образуется белый порошок). для равномерного прокаливания необходимо перемешивать купорос стеклянной палочкой. Следует помнить, что порошок медного купороса сильно раздражает слизистые оболочки. Нужно проводить прокаливание в вытяжном шкафу.
|
|
Пропитка. Смесь компонентов, вхдящих в состав смолы, проводят тщательно, избегая образования пузырьков воздуха. Компоненты отмеряют либо при помощи весов, либо с использованием мерного цилиндра и пипетки в следующем соотношении:
Эпон 812 10 мл (12 г)
HY 964 8 мл (8г)
MNA 4 мл (5г)
катализатор полимеризации DMP30 0,65 мл (0,5г)
Добавлении пластификатора дибутилфталата 1,5-2% от объема смеси позволяет регулировать твердость смолы, что облегчает в последствии ультрамикротомирование.
Затем обезвоженные образцы последовательно инкубируются в растворах смолы с ацетоном в следующих пропорциях:
а) смола: ацетон = 1:3 на 30 мин.
б) смола: ацетон = 2:2 на 30 мин.
в) смола: ацетон = 3:1 на 1-1,5 часа
г) чистая смола на 1-1,5 часа
После этого образцы переносятся в формы для заливки, которые заполняются свежей смолой и выдерживаются в течении ночи при комнатной температуре в закрытом сосуде, для завершения пропитывая образцов. Затем формы помещаются в термостат на 600 на 2-3 для полимеризации. В готовых блоках ясно прослеживается положение образца, что позволяет заточить блок таким образом чтобы на вершине пирамидки оказался исследуемый материал. Горизонтальная плоскость затачивается, как правило, в виде трапеции, что позволяет в дальнейшем определить положение интересующего участка на полутонком срезе.
Микротомирование. Полутонкие срезы толщиной 0,5 мкм, режут на ультрамикротоме стеклянным ножом согласно инструкции к оборудованию, предоставленной производителями. Готовые срезы переносят на предметное стекло в каплю воды и высушивают над спиртовкой или на гистологической плитке до полного испарения воды. Одна из проблем, с которой может столкнуться исследователь на данном этапе, это смывание срезов при дальнейшем окрашивании. Это происходит из-за слабого приклеивания срезов к поверхности предметного стекла, как правило из-за загрязнения стекла, обезжиривание стекол 70% спиртом в течении 15-20 мин, обычно позволяет справиться с этой проблемой, также можно помещать срезы в 10% раствор ацетона, что также способствует лучшемы приклеиванию срезов.
|
|
Срезы окрашиваются 1%-ным раствором метиленового синего (или толуидиного синего, тионином, азур-эозином, гематоксилином и эозином и т.д. в зависимости от задач исследования) с добавлением 1%-ного раствора тетрагидробората натрия в течение 2 минут при нагревании стекла со срезами над спиртовкой. Затем препараты промываются дистилированной водой, подсушиваются и анализируются под световым микроскопом. Представленные виды обзорного окрашивания позволяет определить морфологию клеток, выявить черты характерные для различных стадий клеточного цикла и физиологического состояния клеток. В зависимости от задачи исследования выбирается участок для будущих ультратонких срезов и повторно затачивается пирамидка, вершина которой представляет интересующую область.
Ультрамикротомирование - получение срезов толщиной 50-80нм, выполняют с использованием алмазного ножа, при отсутсвие алмазного ножа, можно использовать стеклянный нож, но в этом случае количество качественных срезов, нарезанных одним ножом, ограниченно. Как правило на одном участке ножа первые 2-3 среза являются наиболее качественными, в дальнейшем качество срезов снижается, на них могут появиться поперечные и продольные полосы, и место на ноже приходится менять. Готовые срезы переносят на специальные медные или никелевые сеточки, предварительно тщательно промытые в ацетоне. Обезжиривание сеточек в органических растворителях способствует плотному и стойкому приклеиванию срезов к поверхности сетки, что влияет на конечное качество электронно-микроскопического препарата.
Контрастирование. Если посмотреть на препараты, не прошедшие этап контрастирования, в электронный микроскоп, то материал будет выглядить бледным, слабо-контрастным, но опытный микроскопист с легкость опознает на срезе основные структуры клетки. Это происходит из-за накопления в компанентах клеток, фиксаторов - глутарового альдегида и четырехокиси осмия, которые кроме фиксирующих свойств обладают контрастирующими. Так фиксация глутар альдегидом хорошо контрастирует хроматин и частично лизосомы, несколько слабее - ядрышко и рибосомы в цитоплазме, умеренный контраст имеют матрикс митохондрий и цитоплазмы. Четырехокись осмия активно реагирует с этиленовыми группами ненасыщенных липидов, фиксируя и контрастируя таким образом мембраны клетки и липосомы. Улучшение контраста может быть достигнуто путем избирательного повышения плотности структур. Для этой цели пригодны контрастирующие вещества с высоким молекулярным весом, лучше всего тяжелые металлы и их соединения. При контрастировании биологического материала увеличение электронной плотности определяется общим числом захваченный тканью атомов или молекул контрастирующего вещества. Таким образом, окончательная электронная плотность зависит от массы молкул (атомов) контрастирующего вещества и от числа захваченных тканью молекул (атомов) этого вещества. К элементам, используемым для контрастирования, относятся: свинец, уран, вольфрам, осмий, марганец, а также иод, железо, труть, барий, ванадий, хром, золото, серебро, никель, лантан, рутений, платина, индий, висмут, стронций и торий. В данном методическом пособии мы остановимся лишь на некоторых общеупотребительных контрастирующих веществах.
|
|
Контрастирование уранилацетатом. Катион уранила связывается с фосфатными и карбоксильными группами, контрастируя практически все структуры клетки. Контрастирование уранилацетатом можно осуществлять после фиксации образцов в четырехокиси осмия, отмытые дистилированной водой образцы инкубируются в течении ночи при 4ОС в водном растворе 2% уранилацетата. Затем промываются 3 раза по 5-10 мин дистилированной водой и подвергаются дегидратации. Также контрастирование уранилацетатом можно проводить на срезах. Водный или спиртовой раствор 1% уранилацетата помещают в виде капли на тефлоновую подложку или или пленку парафилм, сетоку со срезами помещают на каплю и инкубируют в течении 5-15 мин. В зависимости от свежести реактива и образца этот параметр может варьировать. Время инкубации зависит от скорости диффузии уранилацетата в ткани, как было показано экспериментально, за 20 мин катин уранила способен проникнуть в ткань, залитую в эпон, на глубину большую чем толщина ультратонкого среза, поэтому увеличивать время инкубации больше чем на 20 мин не имеет смысла. По оканчании окрашивания сетки тчательно промываются дистилированной водой не менее 2 мин.
|
|
Контрастирование свинцом. Замечательные качества солей свинца как контрастирущего агента были отмечены еще в 1942 году. Использование солей свинца в качестве контрастирующего агента осложняется образованием нестворимого карбоната свинца и других нежелательных осадков, которые выпадают в виде мерких гранул на препаратах, значительно ухудшая их качество. Для преодоления этого недостатка был предложен целый ряд модификаций метода, среди которых наиболее популярна модификация Рейнольдца:
В узкую пробирку с притертой крышкой на 15 мл наливают 6 мл бидистиллированной воды и добавляют 2,28 г. Pb(NO3)2 и 0,325 г. Na3(C6H5O7). Раствор непрерывно перемешивается в течение 30 мин до образования гомогенного белого раствра, затем по каплям, осторожно перемешивая, добавляется 1N NaOH для увеличения pH, пока раствор не станет прозрачным. После этого объем раствора доводится до 10 мл. Важно хранить раствор без доступа воздуха (обычно для этого используется медицинский шприц) при 4ОС.
Контрастирование цитратом свинца проводят на срезах, для этого на тефлоновую подложку или пленку Парафильм помещают капли раствора, обкладывают их гранулами NaOH и закрывают чашкой Петри. Эти предосторожности позволяют снизить содержание СО2 и предотвратить выпадение нерастворимых солей свинца. Продолжительность контрастирования может варьироваться от 3 до 15 мин, и, как правило, подбирается экспериментально. По оканчании окрашивания сетки тчательно промываются дистилированной водой не менее 2 мин. Внимание! Свинец ядовит! Следует избегать вдыхание его паров и попадание в организм через рот и кожу!
Контрастирование рутениевым красным. Сведения о гликокаликсе были существенно дополнены благодаря методу контрастирования рутениевым красным в модификации Люфта. В своих опытах in vitro Люфт доказал, что рутениевый красный реагируют с веществами, которые обладают умеренным или сильным отрицательным зарядом. Это могут быть, главным образом, кислые углеводные соединения, входящие в состав гликокаликса клеток и внеклеточного матрикса (рис. 13). Как предполагают, реакция начинается с образования связи между катионом рутеневого красного и кислым углеводом, при этом субстрат окрашивается в красный цвет, но электронно-микроскопическое контрастирование происходит лишь в процессе обработки четырехокисью осмия, при котором в результате окислительно-востановительной реакции, катализируемой рутениевым красным, низшие окислы осмия осаждаются на окисленном субстрате. Учитывая это, а также в связи
|
|
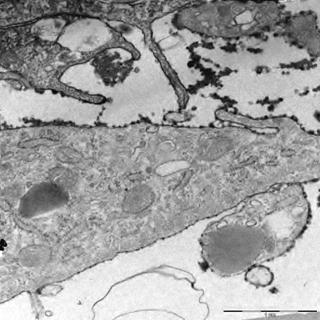 |
Рис. 13. Выявление рутениевым красным кислы х углеводны х соединени й в гликокаликс е клеток и во внеклеточно м матрикс е. Электроннограмма. Бар соответствует 1 мкм.
с тем, что рутениевый красный плохо проникает в биологический материал (мол. вес 858,5, высокий электроположительный заряд) его добавляют непосредственно в глютар альдегидрый фиксатор в концентрации 0,5 мг/мл. В результате использования этого контрастирующего агента, на препаратах хорошо виден гликокаликс клеток, внеклеточный матрикс, а также внутриклеточные кислые мукоиды.
Подготовка опухолевых клеточных линий, зараженных ВБН, для электронномикроскопического анализа
В качестве примера методического подхода подготовки культур клеток для электронно-микроскопического анализа, приведен протокол исследования, целью которого было выявления механизмов взаимодействия ВБН с опухолевыми клетками.
Используемые в работе клеточные линии были посажены на 6-ти луночные планшеты. Через сутки после посадки были заражены ВБН..
Забор материала производили через 3 часа, сутки, 5 дней, 10 или 12 (для ВБН «Адыгея» и «Голубиный» соответственно) после инфекции.
Пробоподготовку проводили по модифицированной нами стандартной методике, что позволило в большей степени сохранить структуру культуральных клеток.
Клетки фиксировали через 3 часа, 1 сутки, 5 суток и 10 суток после обработки вирусом. Фиксацию проводили в 2,5%-ном глутаральдегиде на 0,1 М Na-какодилатном буфере и в 2,5% глутаральдегиде на 0,1 М Na-какодилатном буфере с добавлением рутениевого красного (0,1 М).
Клетки инкубировали с фиксатором 1 час, затем аккуратно снимали их покровным стеклом с лунок и переносили в пробирки.
Пробирки центрифугировали 5 минут при 4200 об./мин., затем убирали фиксатор и заливали клетки 2%-ным раствором агарозы.
С помощью пастеровской пипетки суспензию клеток в агарозе переносили на охлаждающий столик, застывшие агарозные блоки нарезали лезвием на кусочки объемом 1 мм3 .
Полученные агарозные блоки с клетками отмывали 0,1 М Na-какодилатным буфером и проводили дофиксацию 2%-ным раствором OsO4.
Материал, фиксированный без рутениевого красного, инкубировали с 1%-ным водным раствором уранилацетата в течение 12 часов при температуре +4.
Далее материал обезвоживали в серии спиртов восходящей концентрации (30°, 50°, 70°, 90°, 96°), ацетоне и пропитывали смесью ацетона и эпоксидной смолы (3:1, 1:1, 1:3), а затем чистой смолой.
Материал заливали в смесь эпоксидных смол (Epon 812, Epon DDSA, Epon MNA) с катализатором DMP30 и проводили полимеризацию при температуре +60°С в течение 48 часов.
С полученных блоков делали ультратонкие срезы толщиной 50-60 нм на ультрамикротоме Leica UC6 (Германия) и помещали их на никелевые сеточки.
Срезы клеток, предварительно инкубированных с уранилацетатом, контрастировали цитратом свинца по Рейнгольдсу.
Срезы клеток, предварительно инкубированных с рутениевым красным, контрастировали насыщенным спиртовым раствором уранилацетата и цитратом свинца по Рейнгольдсу.
Образцы просматривали под электронным микроскопом Libra 120 (Carl Zeiss, Германия) при ускоряющем напряжении 120 kV.
ЛИТЕРАТУРА
1. Адамс Р., Методы культуры клеток для биохимиков. Москва, Издательство "Мир", 1983, 262 с.
2. Балашов Ю. С., Миккау Н. Е., Изучение живых животных в растровом электронном микроскопе, "Природа", 1977, № 1;
3. Киселев Н. А., Электронная микроскопия биологических макромолекул, М., 1965;
4. Миронов А. А. Методы электронной микроскопии в биологии и медицине. - СПб.: Наука, 1994.
5. Гайер Г. Электронная гистохимия. Москва, Издательство "Мир", 1974, 488
6. Голубев Д. Б., Соминина А. А., Медведева М. Н. Руководство по применению клеточных культур в вирусологии. - Л.: Медицина, 1976, 224 с.
7. Животная клетка в культуре под. ред. Л. П. Дьяконова, В. И. Ситькова. – М.: 2000.
8. Келдыш М.А., Помазков Ю.И. Вирусы, вироиды и микоплазмы растений: К 34 / Учебное пособие.- М.: Изд-во РУДН, 2003 – с. 157
9. Культура животных клеток. Методы. / Под ред. Р. Фрешни. - М.: Мир, 1989, 333 с.
10. Методы общей бактериологии: Пер. с англ./Под ред. Ф. Герхардта и др. — М.: Мир, 1983. — 536 с.
11. Миронов А.А., Комиссарчик Я.Ю., Миронов В.А. Методы электронной микроскопии в биологии и медицине.Спб «Наука» 1994 400 с.
12. Оленов Ю.М., - 1976 - "Новые методы культуры животных тканей".
13. Сергеев В. А. Репродукция и выращивание вирусов животных. - М.: Колос, 1976, 303 с.
14. Синдо Дайзуке. Аналитическая просвечивающая электронная микроскопия. - Москва: Техносфера, 2006.
15. Темников Д.А., Мезина З.Р., Александрова И.С., Ларионова Н.Л. «Основы культивирования клеток животных». Интернет-курс. 2000 – 2003. Доступен на сайте http://www.ksu.ru/nilkto/cell/author.html.
16. Уикли Б., Электронная микроскопия для начинающих.Москва, Издательство "Мир", 324.
17. Уосли Д. Новые методы культуры животных тканей. Перевод с английского Фридлянской И.И., под редакцией Оленова Ю.М., Москва, Издательство "Мир",255.
18. Alberts B., Bray D., Lewis J. et al. Molecular biology of the cell: third edition / Garland Publ. Inc., N.Y. & London. - 1994.
19. Animal Cell Culture: A Practical Approach / Ed. John Masters, 2000.
20. Electron microscopy of enzymes. Principles and methods, v. 1—2, N. Y., 1973—74.
21. Harrison M.A., Rae I.F. General Techniques of Cell Culture, 2001.
22. Freshney R.I. Culture of Animal Cells: A Manual of Basic Technique, 4th Edition, 2000.
23. Langdon S. Cancer Cell Culture: Methods and Protocols, 2003.
24. Tribe М. A., Eraut M. R., Snook R. K., Basic biology course, book 2 — Electron microscopy and cellstructure, Camb., 1975;
25. Для подготовки данной работы были использованы материалы с сайта http://www.study.online.ks.ua/
|
|
|

Состав сооружений: решетки и песколовки: Решетки – это первое устройство в схеме очистных сооружений. Они представляют...

Двойное оплодотворение у цветковых растений: Оплодотворение - это процесс слияния мужской и женской половых клеток с образованием зиготы...

Историки об Елизавете Петровне: Елизавета попала между двумя встречными культурными течениями, воспитывалась среди новых европейских веяний и преданий...

Эмиссия газов от очистных сооружений канализации: В последние годы внимание мирового сообщества сосредоточено на экологических проблемах...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!