

Археология об основании Рима: Новые раскопки проясняют и такой острый дискуссионный вопрос, как дата самого возникновения Рима...

Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...
Археология об основании Рима: Новые раскопки проясняют и такой острый дискуссионный вопрос, как дата самого возникновения Рима...
Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...
Топ:
Основы обеспечения единства измерений: Обеспечение единства измерений - деятельность метрологических служб, направленная на достижение...
Характеристика АТП и сварочно-жестяницкого участка: Транспорт в настоящее время является одной из важнейших отраслей народного...
Эволюция кровеносной системы позвоночных животных: Биологическая эволюция – необратимый процесс исторического развития живой природы...
Интересное:
Аура как энергетическое поле: многослойную ауру человека можно представить себе подобным...
Принципы управления денежными потоками: одним из методов контроля за состоянием денежной наличности является...
Берегоукрепление оползневых склонов: На прибрежных склонах основной причиной развития оползневых процессов является подмыв водами рек естественных склонов...
Дисциплины:
|
из
5.00
|
Заказать работу |
Содержание книги
Поиск на нашем сайте
|
|
|
|
Степень диссоциации: Для количественной характеристики соотношения диссоциированных и недиссоциированных молекул растворенного вещества при данных условиях используют понятие степень диссоциации. Обозначают ее, обычно, буквой «α». Степень диссоциации по своему смыслу ─ это доля распавшихся молекул на ионы:
α=число растворенных молекул, распавшихся на ионы/общее число растворенных молекул
Степень диссоциации зависит и от природы растворителя и растворяемого вещества, температуры и концентрации. С увеличением температуры степень диссоциации увеличивается, так как повышается вероятность разрыва свя-
зей между ионами в молекулах растворенного вещества, и наоборот. С увеличением концентрации растворенного вещества степень диссоциации уменьшается, потому что в растворе появляется значительно количество ионов, и ско-
рость обратного процесса – ассоциации возрастает. Влияние природы растворителя и растворяемого вещества на степень диссоциации связано с характером собственных связей и их энергией в молекулах веществ и взаимодействием молекул между собой.
По величине степени диссоциации все электролиты условно подразделяют на 3 группы: растворы, в которых определяемая (кажущаяся) степень диссоциации растворенного вещества составляет величину большую 30% (α > 30%) называют сильными электролитами, со степенью диссоциации α меньше 3% (α < 3%) – слабыми электролитами, растворы со степенью диссоциации растворенного вещества в промежуточной области α = 3 ÷ 30% называют средними электролитами.
К сильным электролитам в водном растворе относятся почти все соли, многие неорганические кислоты (HCl, H2SO4, HBr, HI, HNO3 и др.) и гидроксиды щелочных и щелочноземельных металлов (NaOH, KOH, Ba(OH)2 и др.).
К слабым электролитам относятся водные растворы H2S, HCN, H2SiO3, H3BO3, гидроксиды многих металлов (Cu(OH)2, Fe(OH)3, Zn(OH)2, Ni(OH)2, Pb(OH)2, Al(OH)3 и многие, многие другие), органические кислоты, соли ртути HgCl2, CdCl2 и некоторые другие.
Средними электролитами принято считать водные растворы фосфорной, сернистой, муравьиной, щавелевой и некоторых других.
Константа диссоциации: применяется для характеристики слабых электролитов. Вследствии того, что слабые электролиты диссоциируют на ионы не полностью, в их растворах устанавливается динамическое равновесие между недиссоциированными молекулами и ионами. Для слабого электролита общей формулы AnBm уравнение диссоциации имеет вид: AnBm  nAm++mBn-. Применяя закон действующих масс выражение константы равновесия:
nAm++mBn-. Применяя закон действующих масс выражение константы равновесия: 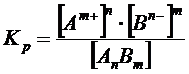 , где
, где 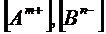 - равновесные концентрации ионов Am+ и Bn-,
- равновесные концентрации ионов Am+ и Bn-, 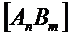 - равновесная концентрация молекул AnBm.
- равновесная концентрация молекул AnBm.
Константу равновесия в этом случае называют константой диссоциации или константой ионизации. Константа ионизации характеризует способность электролита диссоциировать на ионы. Чем больше константа диссоциации, тем легче электролит распадается на ионы, тем больше ионов в его растворе, тем сильнее электролит. Для слабого электролита константа диссоциации – постоянная величина при данной температуре, которая не завсит от концентрации раствора. Константа диссоциации зависит от природы электролита, природы растворителя и температуры.
Поскольку константа равновесия относится к процессу диссоциации, то ее можно назвать константой диссоциации Кд. Она показывает, что в состоянии равновесия концентрации образовавшихся ионов и нераспавшихся молекул находятся во взаимосвязи. Изменение концентрации любого из компонентов в растворе, автоматически приведет к изменению концентрации других, но так, чтобы их соотношение оставалось тем же самым – равным константе диссоциации Кд = Кр. Если константа диссоциации больше единицы (числитель выражения больше знаменателя), то в растворе очень мало нераспавшихся молекул и много ионов. Следовательно, чем меньше константа диссоциации, тем слабее диссоциирует растворенное вещество, тем прочнее его молекулы в данном растворителе.
Связь константы равновесия со степенью диссоциации:
После упрощения получим: Кд= α2C/1−α. Полученное выражение носит название закона разбавления В. Оствальда. Оно связывает степень диссоциации с концентрацией растворенного вещества. Для слабых электролитов, у которых степень диссоциации меньше 0,03 (α < 0,03), величиной α в знаменателе этого выражения можно пренебречь, так как 0,03 много меньше единицы (0,03 << 1). Тогда уравнение В. Оствальда можно упростить: Кд ≅ α 2С или α≅  . Теперь явно видно, что степень диссоциации обратно пропорциональна концентрации растворенного вещества: чем выше концентрация, тем меньше степень диссоциации. Слабые многоосновные кислоты, а также двух- и более кислотные основания характеризуются несколькими константами диссоциации, соответствующими каждой ступени диссоциации. Сильные же электролиты, у которых молекулы растворенного вещества практически все полностью диссоциированы, не подчиняются закону действующих масс.
. Теперь явно видно, что степень диссоциации обратно пропорциональна концентрации растворенного вещества: чем выше концентрация, тем меньше степень диссоциации. Слабые многоосновные кислоты, а также двух- и более кислотные основания характеризуются несколькими константами диссоциации, соответствующими каждой ступени диссоциации. Сильные же электролиты, у которых молекулы растворенного вещества практически все полностью диссоциированы, не подчиняются закону действующих масс.
Основные представления теории растворов сильных электролитов. Истинная и кажущаяся степень диссоциации в растворах. Концентрация и активность ионов. Коэффициент активности. Ионная сила растворов электролитов.
В сильных электролитах растворенное вещество практически диссоциировано нацело (степень диссоциации ≈ 100 %). Так, например, в водном растворе поваренной соли нельзя обнаружить отдельную молекулу NaCl, а имеются
отдельные гидратированные ионы натрия Na+ и хлора Cl-. Следовало бы для такого раствора ожидать величину изотонического коэффициента равной двум (i=2), что соответствует распаду молекулу поваренной соли на 2 иона. Но фактически изотонический коэффициент меньше двух и зависит от концентрации. Причем, чем меньше концентрация, тем ближе значение изотонического коэффициента к теоретической величине – двум.
Факт полной диссоциации растворенного вещества в растворах сильных электролитов на ионы нельзя отрицать. Так почему же свойства растворов (осмотическое давление, электропроводность, температуры начала кипения и замерзания растворов и др.) не соответствуют концентрациям ионов в растворе? Чтобы ответить на этот вопрос, рассмотрим зависимость, например, электропроводности от концентрации растворенного вещества (той же поваренной соли).
Для определения электропроводности раствора, находящегося в сосуде, нужно в него ввести электроды А (положительный электрод) и К (отрицательный электрод).
Измеряя в цепи силу тока амперметром, зная разность потенциалов на электродах, их площадь и расстояние между ними, можно рассчитать электропроводность (величину обратную сопротивлению) раствора.
Представим себе, что мы в сосуд с чистой водой начали добавлять по одной молекуле поваренной соли NaCl. Молекулы поваренной соли сразу же диссоциируют на ионы. В растворе начинает увеличиваться концентрация заряженных частиц – носителей зарядов. Чем их больше, тем выше электропроводность. Заметим при этом, что ионы в растворе движутся к электродам независимо друг от друга, так как их очень мало. По мере увеличения концентрации растворенных молекул NaCl пропорционально (линейно) должна увеличивается и электропроводность раствора.
При дальнейшем увеличении концентрации поваренной соли, а, следовательно, и ионов, последних в растворе окажется так много, что они будут находиться в непосредственной близости друг от друга и начнут взаимодействовать между собой. Доля этого взаимодействия будет усиливаться с ростом концентрации.
При этом каждый ион в растворе окажется окруженным роем противоположно заряженных ионов. Двигаться такому рою в растворе под действием внешнего электрического поля будет сложно. Мало того, что размеры такого ассоциата возросли во много раз по сравнению с отдельным ионом, так
они еще и движутся в разных направлениях (к разным электродам). Такое взаимодействие ионов друг с другом и растворителем влияет на скорость их движения в электрическом поле, что ведет в конечном итоге к понижению электропроводности с увеличением концентрации. Понятно, что эти процессы не связаны с изменением истинной степени диссоциации α растворенного вещества, как у слабых электролитов, а обуславливаются взаимодействием ионов между собой, образованием ассоциатов из ионов. Поэтому, определяя электропроводность концентрированного раствора сильного электролита или любое другое свойство, зависящее от концентрации частиц, мы находим его лишь как функцию кажущейся концентрации частиц и соответственно кажущейся степени диссоциации сильного электролита.
Свойство раствора проявляется так, как если бы концентрация ионов в растворе оказалась меньше, чем это соответствует их аналитической концентрации при условии полной диссоцииации растворенного вещества. Кажущаяся
концентрация соответствует тому или иному свойству, но не отражает истинную аналитическую концентрацию частиц в концентрированных растворах, что и обуславливает неприменимость закона действующих масс, оперирующего
истинными концентрациями частиц.
Кажущаяся концентрация выступает в роли активной (эффективной), действующей концентрации, оответствующей тому или иному свойству раствора. При использовании величины активной концентрации в соответствующих уравнениях (закона действующих масс, Рауля, Вант Гоффа и др.), они становятся справедливыми при любых концентрациях. Активную концентрацию называют активной концентрацией, или просто активностью, и обозначают
буквой «а». Она связана с истинной аналитической концентрацией С простым соотношением а = f C, где f – коэффициент активности. Коэффициент активности f формально учитывает (а соответственно и активная концентрация) все виды взаимодействий частиц в растворе, приводящие к отклонению свойств растворов от идеальных. Его определяют опытным путем, измеряя экспериментальную величину какого-либо свойства раствора и сравнивая ее с теоретическим значением
f =Экспериментальная величинаа свойства/теоретическое значение свойства.
Числитель и знаменатель имеют одну и ту же размерность, поэтому коэффициент активности ─ безразмерная величина. Формально коэффициент активности показывает, во сколько раз отличается активная концентрация от истинной ана-
литической концентрации в растворе.
Для предельно разбавленных растворов, где практически отсутствует взаимодействие ионов между собой из за их удаленности друг от друга, коэффициент активности равен f =1. И тогда активная концентрация и аналитическая совпадают: а = С. Активная концентрация также выражается в моль/л, но является как бы действующей, эффективной концентрацией.
Введение понятия активной концентрации является лишь удобным формальным приемом, позволяющим рассчитывать свойства растворов при высоких концентрациях. Использование этой величины говорит о том, что в раство-
ре существуют какие-то процессы взаимодействия частиц между собой, конкретный механизм которого не раскрывается.
Cледует отметить, что значение коэффициента активности для данного раствора зависит, очевидно, от способа выражения концентрации растворенного вещества. Поскольку обычно для характеристики раствора используются молярная доля (χВ), молярная (СМ) и моляльная (Сm) концентрации, то и используются три различных типа коэффициентов активности.
Ионная сила (ионность) раствора: Использование понятия активной концентрации не совсем правомерно. В
растворе сильных электролитов молекул как таковых практически нет. Свойства растворов обуславливают ионы. Следовательно, нужно говорить не об активности (активной концентрации) молекул, а об активности ионов: активной
концентрации положительных ионов а+ и активной концентрации отрицательных ионов а-, и соответственно ─ о коэффициентах активности положительных и отрицательных ионов f+ и f-, поскольку иметь дело с одним видом
ионов в растворе невозможно.
Активная концентрация иона пропорциональна его концентрации в растворе: a+ = f+ C+ и a- = f- C-.
В водных растворах коэффициент активности данного вида ионов f+ и f- зависит от природы иона (заряда, радиуса), температуры и концентрации всех ионов, присутствующих в растворе. Коэффициенты активности ионов изменяются в
очень широких пределах: в области разбавленных растворов они стремятся к единице, а в области концентрированных растворов могут достигать десятков и даже сотен единиц. В разбавленных растворах (меньше 0,1 моль/л) коэффици-
енты активности ионов зависят главным образом от концентрации и заряда ионов, присутствующих в растворе, и мало зависят от природы растворенных веществ.
Суммарное электростатическое взаимодействие между всеми ионами раствора принято характеризовать ионностью раствора, или ионной силой, обозначаемой обычно I. Если наряду с ионами данного электролита в растворе
присутствуют и другие ионы второго электролита, то ионность среды I будет складываться из интенсивностей отдельных полей всех ионов обоих электролитов.
Теория и опыт показывают, что вклад каждого вида ионов в общую интенсивность электрического поля (ионность раствора) пропорционален концентрации этих ионов C и квадрату заряда z иона: I ≈ Z2 C.
Для водных растворов коэффициент пропорциональности в этом выражении равен 0,5. И тогда ионность раствора, содержащего различные ионы с соответствующими зарядами и концентрациями, определится следующим выражени-
ем: I = 0,5(z12 C1 + z22 C2 + z32 C3 + …zn2 C n) = 0,5∑ zi2 Ci.
Для разбавленных водных растворов электролитов (с ионностью I < 0,01) коэффициенты активности отдельных ионов могут быть рассчитаны из выражения Дебая ─ Хюккеля: lg fi = - 0,51 Zi 2√ I, где fi, zi –коэффициент активности и заряд i – го иона (катиона или аниона).
По найденным коэффициентам активности отдельных ионов рассчитываются их активные концентрации ионов (а+ и а-), а затем и активная концентрация растворенного вещества а.
Средние коэффициенты активности ионов f+ электролитов также могут быть рассчитаны из уравнения Дебая –Хюккеля, если представить его в более
общем виде: lg f+ = - A ⋅ Z+⋅ Z- √ I, где А – коэффициент, зависящий от природы растворителя и температуры (для
водных растворов он равен А = 0,51 при 298 К).
Найденные таким образом активные концентрации можно использовать для расчета свойств растворов и констант равновесия любого обратимого процесса (в том числе и диссоциации), протекающего в растворе.
На нескольких примерах рассмотрим расчет ионности среды I, активной концентрации а и коэффициентов активности f.
Таким образом, представление об идеальных растворах – это упрощение, хотя и очень полезное. Ближе всего к этому идеалу стоят растворы, состоящие из неполярных и слабо поляризуемых молекул, например, углеводородов. В растворах с полярными молекулами и тем более с ионами очень велико взаимодействие частиц, и они не могут считаться полностью свободными.
Значит ли это, что у реальных растворов не будут проявляться коллигативные свойства? Конечно же, будут, и обычно даже сильнее, чем у идеальных. Но они не будут точно описываться такими простыми уравнениями, как у идеальных растворов. Например, если растворенное вещество сильно сольватируется, то есть связывает молекулы растворителя, то давление пара растворителя будет понижено сильнее, чем ожидается по закону Рауля. В этом случае говорят об отрицательном отклонении от идеальности. Гораздо реже встречается положительное отклонение, когда реальное давление пара больше идеального, то есть молекулы как бы выталкиваются из раствора. Не только закон Рауля, но и все родственные ему количественные закономерности, в частности, закон действия масс, тоже перестают быть точными для неидеальных растворов, если в них использовать обычные концентрации. Поэтому введено понятие активности. Активность – это термодинамическая величина, имеющая смысл “действующей концентрации”, то есть такая величина, которую нужно подставлять вместо концентраций в термодинамические уравнения для идеальных растворов, чтобы они точно выполнялись и для реальных растворов. Очень важно, что активности, определенные из одних экспериментов (например, через давление пара), применимы и для описания других свойств, например, химических равновесий в растворах. Это доказывает, что они имеют реальный смысл. Коэффициент активности f – это отношение активности к концентрации. Для идеальных растворов он по определению равен единице, для реальных – обычно меньше единицы. Это отражает тот факт, что из-за межмолекулярных и межионных взаимодействий частицы не вполне свободны и ведут себя так, будто их концентрация несколько меньше фактической.
Далее мы будем для простоты везде пользоваться концентрациями (считая все f = 1), но не надо забывать, что без учета коэффициентов активности расчеты химических равновесий не могут быть точными. Поэтому основное внимание будем обращать на качественную сторону явлений, а равновесные концентрации вычислять с точностью не более 1-2 значащих цифр.
Кислоты, основания, амфотерные гидроксиды, соли с точки зрения теории электролитической диссоциации. Диссоциация солей средних, кислых, основных. Ступенчатая диссоциация. Теории кислот и оснований Аррениуса, Бренстеда, Льюиса.
Кислоты – электролиты, которые при диссоциации образуют только один вид катионов – катионы водорода H+.
Слабые многоосновные кислоты диссоциируют ступенчато. Число ступеней диссоциации зависит от основности слабой кислоты. Растворы кислот имеют некоторые общие свойства, которые, согласно теории электролитической диссоциации, объясняются присутствием в их растворах гидратированных ионов водорода H+ (H3O+).
Основания – электролиты, которые при диссоциации образуют только один вид анионов – гидроксид-ионы OH-.
Слабые многокислотные основания диссоциируют ступенчато. Число ступеней диссоциации определяется кислотностью слабого основания. Основания имеют некоторые общие свойства. Общие свойства оснований обусловлены присутствием гидроксид-ионов OH-.
Каждая ступень диссоциации слабых многоосновных кислот и слабых многокислотных оснований характеризуется оперделенной константой диссоциации: К1, К2, К3, причем  . Это объясняется тем, что энергия, которая необходима для отрыва иона H+ или OH- от нейтральной молекулы кислоты или основания минимальна. При диссоциации по следующей ступени энергия увеличивается, потому что отрыв ионов происходит от противоположно заряженных частиц.
. Это объясняется тем, что энергия, которая необходима для отрыва иона H+ или OH- от нейтральной молекулы кислоты или основания минимальна. При диссоциации по следующей ступени энергия увеличивается, потому что отрыв ионов происходит от противоположно заряженных частиц.
Амфотерные гидрооксиды – это слабые электролиты, которые при диссоциации образуют одновременно катионы водорода и гидроксид анионы, т.е. диссоциируют по типу кислоты и по типу основания. К ним оносятся: Be(OH)2, Zn(OH)2, Sn(OH)2, Al(OH)3, Cr(OH)3 и вода.
В амфотерных гидроксидах диссоциация по типу кислот и по типу оснований происходит, потому что прочность химических связей между атомами кислорода и водорода почти одинаковая. Поэтому в водном растворе эти связи разрываются одновременно, и амфотерные гидрооксиды при диссоциации образуют катионы H+ и анионы OH-.
Нормальные соли – сильные электролиты, образующие при диссоциации катионы металла и анионы кислотного остатка.
Кислые соли – сильные электролиты, диссоциирующие на катимон металла и сложный анион, в состав которого входят атомы водорода и кислотный остаток.
Основные соли – электролиты, которые при диссоциации образуют анионы кислотного остатка и сложные катионы, состоящие из атомов металла и гидроксогрупп OH-.
Согласно представлениям С. Аррениуса кислота есть всякое водородосодержащее соединение, которое в водном растворе при диссоциации образует ион водорода Н+, как, например:
HCl → H+ + Cl-,
HNO3 → H+ + NO3-,
H2SO4 → 2H+ + SO42-.
А основание – всякое гидроксилсодержащее соединение, которое в водном растворе при диссоциации дает ион гидроксила ОН-, например:
NaOH → OH- + Na+,
Ba(OH)2 → 2OH- + Ba2+.
Процесс нейтрализации кислоты основанием заключается во взаимодействии ионов водорода Н+ и гидроксила ОН- с образованием воды: Н+ + ОН- → Н2О.
Заметим, что данные модельные представления С. Аррениуса о кислотах и основаниях не являются универсальными. Они применимы лишь к водным растворам, так как хорошо объясняют их электропроводность, каталитические свойства кислот (вследствие большой подвижности ионов водорода). А также факт примерно одного и того же значения теплового эффекта реакции нейтрализации различных кислот основаниями, равным ∆Н = -58 кДж/моль, так как в основе лежит одна и та же реакция ─ соединение иона водорода с ионом гидроксила, и становятся понятными многие другие стороны поведения кислот, оснований.
Теории кислот и оснований Аррениуса, Бренстеда, Льюиса: Любое вещество в определенных условиях может проявлять свойства кислоты и основания по отношению к какому-либо другому веществу, включая и растворитель. Со времен Аррениуса, по определению которого кислоты в водных растворах диссоциирует на ионы водорода и анионы, а основания диссоциируют на гидроксид-ионы и катионы, круг веществ, участвующих в реакциях кислотно-основного равновесия, значительно расширился. Общепринятой считается протонная теория Бренстеда–Лоури. Протонная теория Бренстеда–Лоури применима лишь к протонодержащим или протон-присоединяющим веществам. Согласно этой теории кислотой называется вещество, способное быть донором протонов, а основанием – вещество, которое может присоединить (акцептировать) протон:
HAn ↔ An– + Н+. По определению, HAn – кислота, An– – основание, сопряженное с этой кислотой. Любой кислоте соответствует сопряженное с ней основание. В определенных условиях многие вещества могут вести себя как кислота или как основание в зависимости от того, с каким другим веществом оно вступает в реакцию. Эти два понятия неразделимы, а потому правильнее говорить о кислотно-основных свойствах данного вещества.
В дальнейшем появилось много других экспериментальных данных, связанных в основном с неводными растворителями, и соответственно новых концепций понимания кислот и оснований. Классифицировать подобные взаимодействия позволила электронная теория кислот и оснований Льюиса.
Электронная теория Льюиса допускает, что участие в кислотно-основном равновесии протона необязательно, поэтому ее называют апротонной. Согласно апротонной (электронной) теории, кислотой называется вещество, способное присоединять электронную пару, а основанием – вещество, способное отдавать электронную пару. Основанием Льюиса могут быть анионы (Cl-, OH-) или нейтральные молекулы (H2O, NH3). Кислотами Льюиса могут быть катионы (H+, Fe2+) или нейтральные молекулы, имеющие свободные валентные орбитали (BF3, AlCl3, ZnCl2)
При взаимодействии донора электронной пары:NН3 (основание) и акцептора электронной пары НCl (кислота) образуется более устойчивое электронное окружение (октет) за счет донорно-акцепторной (двухэлектронной двухцентровой) связи.
Ступенчатая диссоциация:
Многоосновные кислоты и многокислотные основания диссоциируют ступенчато. Так, например, молекулы угольной кислоты Н2СO3 вначале диссоциируют с отще-
плением одного иона водорода по уравнению
1) H2СO3↔ H+раствор + [HСO3]-раствор.
Степень диссоциации этого процесса α1. Затем образовавшиеся кислые анионы угольной кислоты [HСO3]- в свою очередь распадаются под действием молекул воды на более простые ионы:
2) [HСO3]- ↔ H+раствор + [СO3]2-раствор
с отщеплением второго иона водорода. Но этот процесс идет уже много труднее, так как иону водорода Н+ приходится отрываться уже от заряженной отрицательно частицы, а не от нейтральной молекулы, как в первом случае. Поэтому степень диссоциации второй ступени α2 характеризуется меньшим значением, т.е. α1>α2. Вследствие этого в растворе содержится лишь небольшое число
ионов [СO3]2-.
Фосфорная кислота диссоциирует в три ступени:
1) Н3РО4 ↔ Н+ + [Н2РО4]-,
2) [H2PO4]- ↔ H+ + [HPO4]2-,
3) [HPO4]2- ↔H+ + [PO4]3-,
каждая из них так же характеризуется соответствующей степенью диссоциации: α1, α2 и α3, которые тоже находятся в следующем соотношении: α 1> α 2 > α 3.
А это означает, что ионов [PO4] 3- в растворе почти нет.
Основания, содержащие более одной гидроксильной группы в молекуле, также диссоциируют ступенчато. Например, гидроксид кальция Са(ОН)2 диссоциирует в две стадии:
1) Са(ОН)2 → [CaOH]+ + OH- (α1)
2) [CaOH] + → Ca 2++ OH - (α2).
Гидроксиды многих металлов (Zn(OH)2, Al(OH)3, Be(OH)2, Cr(OH)3, Pb(OH)2 и других) могут диссоциировать и по кислотному, и по основному механизму.
Например, диссоциацию гидроксида цинка Zn(OH)2 можно выразить уравнениями:
1) Zn(OH)2 ↔ [Zn(OH)]+ + OH-,
2) H2ZnO2 ↔ [HZnO2]+ + H+ или
[Zn(OH)]+ + OH- ↔ Zn(OH)2 ↔ H2ZnO2 ↔ [HZnO2]- + H+.
Диссоциация по тому или иному пути определяется в зависимости от среды: в кислой среде, где концентрация ионов водорода Н+ велика, ионы гидроксила
связываются с ионами водорода, смещая равновесие в сторону образования ионов [Zn(OH)]+, а в щелочной (концентрация ОН- значительна) ионы ОН-, взаимо-
действуя с ионами водорода, смещают равновесие в сторону образования ионов [HZnO2]-. Так или иначе оба процесса приводят к образованию слабого электролита – воды:
Н+ + ОН- = Н2О.
Соли, как известно, являются продуктом реакции нейтрализации. В зависимости от полноты нейтрализации соли могут быть: средние, или нейтральные
(например, К3РО4, Са(NO3)2, NaCl); кислые, если не все водородные ионы в молекуле кислоты замещены на металл (например, КН2РО4, NaHSO4, СsHCO3), и основные, если в составе молекулы соли имеется гидроксильная группа ОН- (например, ZnOHCl, Fe(OH)2Cl, AlOHSO4). В водных растворах они диссоциируют на катионы металлов (или комплексные катионы) и анионы (одноатомные или
многоатомные). Средние соли называют используя название кислотного остатка в именительном падеже и катиона металла в родительном. Например, соли К3РО4, Са(NO3)2, NaCl называют фосфат калия, нитрат кальция, хлорид натрия соответственно. Кислые соли называют аналогичным образом, добавляя к названию кислотного остатка приставку гидро, дигидро, тригидро, и.т.д. Например, соли КН2РО4, NaHSO4, СsHCO3 называют дигидрофосфат калия, гидросульфат натрия, гидрокарбонат цезия соответственно.
Основные соли называют аналогично средним, добавляя к названию катиона приставку гидроксо, дигидроксо и. т.д. Например, соли ZnOHCl, Fe(OH)2Cl, AlOHSO4 называют хлорид гидроксоцинка, хлорид дигидроксожелеза, сульфат гидроксоалюминия соответственно. Средние соли диссоциируют практически полностью. Например, в водном растворе сульфата натрия Na2SO4 практически нет молекул Na2SO4, а имеются только ионы Na+ и SO42-, что выражается уравнением Na2SO4 → 2 Na+ + SO42-.
Кислые соли диссоциируют ступенчато, отщепляя вначале ионы металлов, а затем ионы водорода, например:
1) KН2РО4 → К+ + [Н2 РО4]-,
2) [H2PO4]- ↔ H+ + [HPO4]2-,
3) [HPO4]2- ↔ H+ + [PO4]3-.
Ступенчато диссоциируют также и основные соли. Например, дигидроксохлорид железа Fe(OH)2Cl диссоциирует в три стадии, которые можно предста-
вить следующими уравнениями:
1) Fe(OH)2Cl → [Fe(OH)2]+ + Cl-,
2) [Fe(OH)2]+ ↔ [Fe(OH)]2+ + OH-,
3) [Fe(OH)]2+ ↔ Fe3+ + OH-.
Диссоциация кислых и основных солей протекает практически полностью по первой ступени. На тех стадиях, когда происходит распад сложных анионов кислых солей или катионов основной соли (появляются ионы водорода
или гидроксила) степень диссоциации очень незначительна. А это значит, что в растворе практически отсутствуют ионы РО43- и Fe3+ при ступенчатой диссоциации дигидрофосфата калия и дигидроксохлорида железа соответственно в при-
веденных примерах.
Реакции ионного обмена в растворах. Обратимые и необратимые реакции, признаки
необратимости реакций.
Реакции ионного обмена - это реакции между ионами, образовавшимися в результате диссоциации электролитов.
Согласно теории электролитической диссоциации все реакции в водных растворах электролитов являются реакциями между ионами. Они называются ионными реакциями, а уравнения этих реакций - ионными уравнениями. Они проще уравнений реакций, записанных в молекулярной форме, и имеют более общий характер.
При составлении ионных уравнений реакций следует руководствоваться тем, что вещества малодиссоциированные, малорастворимые (выпадающие в осадок) и газообразные записываются в молекулярной форме. Знак ↓, стоящий при формуле вещества, обозначает, что это вещество уходит из сферы реакции в виде осадка, знак ↑ обозначает, что вещество удаляется из сферы реакции в виде газа. Сильные электролиты, как полностью диссоциированные, записывают в виде ионов. Сумма электрических зарядов левой части уравнения должна быть равна сумме электрических зарядов правой части.
Ионными уравнениями могут быть изображены любые реакции, протекающие в растворах между электролитами. Если при таких реакциях не происходит изменения зарядов ионов (не изменяется степень окисления), то они называются ионообменными.
Правила составления ионных уравнений реакций:
1. Нерастворимые в воде соединения (простые вещества, оксиды, некоторые кислоты, основания и соли) не диссоциируют.
2. В реакциях используют растворы веществ, поэтому даже малорастворимые вещества находятся в растворах в виде ионов.
3. Если малорастворимое вещество образуется в результате реакции, то при записи ионного уравнения его считают нерастворимым.
4. Сумма электрических зарядов ионов в левой и в правой части уравнения должна быть одинаковой.
Порядок составления ионных уравнений реакции:
1. Записывают молекулярное уравнение реакции
MgCl2 + 2AgNO3 = 2AgCl + Mg(NO3)2
2. Определяют растворимость каждого из веществ в воде с помощью таблицы растворимости
pастворим MgCl2
pастворимо AgNO3
нерастворим AgCl
pастворим Mg(NO3)2
3. Записывают уравнения диссоциации растворимых в воде исходных веществ и продуктов реакции:
MgCl2 = Mg2+ + 2Cl-
AgNO3 = Ag+ + NO3-
Mg(NO3)2 = Mg2+ + 2NO3-
4. Записывают полное ионное уравнение реакции
Mg2+ + 2Cl- + 2Ag+ + 2NO3- = 2AgCl + Mg2+ + 2NO3-
5. Составляют сокращенное ионное уравнение, сокращая одинаковые ионы с обеих сторон:
Mg2+ + 2Cl- + 2Ag+ + 2NO3- = 2AgCl + Mg2+ + 2NO3-
Ag+ + Cl- = AgCl
Условия необратимости реакций ионного обмена
1. Если образуется осадок (↓) (смотри таблицу растворимости)
Pb(NO3)2 + 2KI = PbI2↓ + 2KNO3
Pb2+ + 2I- = PbI2↓
2. Если выделяется газ (↑)
Na2CO3 + H2SO4 = Na2SO4 + H2O + CO2↑
CO32- + 2H+ = H2O + CO2↑
3. Если образуется малодиссоциированное вещество (H2O)
Ca(OH)2 + 2HNO3 = Ca(NO3)2 + 2H2O
H+ + OH- = H2O
3а. Если образуются комплексные соединения (малодиссоциированные комплексные ионы)
CuSO4 • 5H2O + 4NH3 = [Cu(NH3)4]SO4 + 5H2O
Cu2+ + 4NH3 = [Cu(NH3)4]2+
|
|
|

Особенности сооружения опор в сложных условиях: Сооружение ВЛ в районах с суровыми климатическими и тяжелыми геологическими условиями...
Биохимия спиртового брожения: Основу технологии получения пива составляет спиртовое брожение, - при котором сахар превращается...

Опора деревянной одностоечной и способы укрепление угловых опор: Опоры ВЛ - конструкции, предназначенные для поддерживания проводов на необходимой высоте над землей, водой...

Индивидуальные очистные сооружения: К классу индивидуальных очистных сооружений относят сооружения, пропускная способность которых...
© cyberpedia.su 2017-2026 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!

