Лечение обычно не требуется, необходимо потреблять достаточно глюкозы и воды, чтоб восполнить их потерю.
NB! – настороженность в отношении сахарного диабета.
Таблица 3 – Характеристика ренальной глюкозурии
| Клиника
| Лабораторные данные
| Дифференциальный
диагноз
| Лечение
|
| Слабость, чувство голода, жажды, полиурия. Задержка физического
развития.
| Глюкозурия.
Суточная экскреция глюкозы с мочой составляет от 2 до 100г.
| При исключении других тубулопатий, обращает внимание на отсутствие экскреции с мочой аминокислот, элект-ролитов, бикар-бонатов КОС в норме, рН мочи в норме.
Сахарный диабет характеризуется повышением уровня сахара в крови.
| Избегать перегрузки углеводами. При развитии гипогли-кемии возможно дополнительное введение глюкозы 5-10% р-р в/в капельно.
|
Аминоацидурии
В проксимальных канальцах идентифицированы транспортные системы нескольких аминокислот.
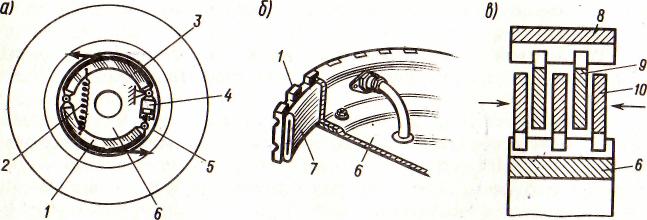
Реабсорбция аминокислот происходит также по механизму сопряженного с Na+ транспорта. Аминокислоты, как и глюкоза, в 95% полностью реабсорбируются эпителиоцитами проксимального канальца. Этот процесс осуществляется с помощью вторично-активного транспорта, то есть энергия идет на работу натриевого насоса. Выделяют не менее 4 транспортных систем для переноса различных аминокислот (нейтральных, двуосновных, дикарбоксильных и аминокислот). Эти же системы транспорта действуют и в кишечнике для всасывания аминокислот (рис.4).

Рисунок 4 - Транспорт аминокислот (4)
Каждый из механизмов относительно специфичен и обеспечивает реабсорбцию аминокислот одной группы. В частности, механизм реабсорбции двуосновных аминокислот участвует во всасывании лизина, аргинина, орнитина и, возможно, цистина. При введении в кровь одной из аминокислот указанной группы в кровь и создании избыточной ее концентрации в плазме крови, начинается усиленная экскреция почкой аминокислот именно этой группы. Транспорт отдельных групп аминокислот управляется специфичными им генетическими механизмами. Описаны наследственные заболевания, одним из проявлений которых служит увеличенная экскреция определенных групп аминокислот (аминоацидурия). Нарушения могут быть специфическими (например, двухосновные аминокислоты при цистинурии) или генерализованными. Аминоацидурии могут возникнуть в результате генетических дефектов в одном из нескольких системах транспорта аминокислот в проксимальных канальцах. С клинической точки зрения целесообразно классифицировать транспортные системы на три большие категории: кислые, основные и нейтральные транспортеры (табл. 4).
Таблица 4 - Классификация наследственных аминоацидурий
| Основные категории
| Формы
| Связанные аминокислоты
|
| Кислые аминокислоты
| Кислотная аминоацидурия
| Глутамат, аспартат
|
| Основные аминокислоты и цистин
| Цистинурия
Непереносимость лизинурического белка Изолированная цистинурия
Лизинурия
| Цистин, лизин, аргинин, орнитин
Лизин, аргинин, орнитин
Цистин
Лизин
|
| Нейтральные аминокислоты
| Болезнь Хартнупа
«Синдром голубых пеленок»
Иминоглицинурия
Глицинурия
Метионинурия
| Аланин, аспарагин, глутамин, гистидин, изолейцин, лейцин, фенилаланин, серин, треонин, триптофан, тирозин, валин
Триптофан
Глицин, пролин, гидроксипролин
Глицин
Метионин
|
Кислая аминоцидурия (также известная как дикарбокси-аминоацидурия) затрагивает транспорт глютамата и аспартата. Она возникает вследствие дефекта ЕААС1 – натрий-калий-зависимого глютамат транспортера с высоким сродством. Клинических проявлений не имеет, прогноз благоприятный.
Описаны четыре синдрома, обусловленные дефектом транспорта основных аминокислот и/или цистина: цистинурия, лизинурическое нарушение толерантности к белку, изолированная цистинурия и изолированная лизинурия.
Основная аминоацидурия
В данную группу входят первичные тубулопатии с ведущим синдромом нефролитиаза (табл. 5).
Таблица 5 - Первичные тубулопатии с ведущим синдромом нефролитиаза
|
| Цистинурия
| Глицинурия
| Аминоглицинурия
|
| Тип
наследо-
вания
| Аутосомно-рецессивный тип. Частота 1:20.000
| Доминантный, сцепленный с Х-хромосомой, болеют только девочки
| Аутосомно-рецессивный тип
|
| Патоге-нез
| Нарушение транспорта цистина в слизистой оболочке тонкой кишки и в почечных канальцах. Выделяют 3 типа;
1-й тип. Отсутствие транспорта цистина и диаминомонокарбо-натовых аминокислот в кишечнике и почках.
2-й тип. Снижение до 50% транспорта цистина в почках и полное отсутствие транспорта диаминомонокарбоновых аминокислот в почках и кишечнике
3-й тип: снижение транспорта этих аминокислот в почках при норме всасывания их в кишечнике
| Нарушение реабсорбции глицерина в проксимальных канальцах
| Нарушение канальцевого транспорта пролина, гидросипролина, глицерина
|
| Клиника
| Клинические признаки чаще с 10-20л:
-почечная колика;
-боли в животе;
- нарушение уродинамики;
- артериальная гипертензия;
- отставание в физическом развитии
| Количественное определение глицина в моче и крови
| Возможно отсутствие клинических признаков и в клинике:
-признаки нефролитиаза;
-умственная отсталость;
- неврологическая симптоматика
- снижение слуха
|
| Диагноз
| Параклиника: кристаллы цистина в моче, выявление аминоацидурии при хромотография мочи
| Количественное определение глицина в моче и крови
|
|
| Диффе-
ренциаль
ный диагноз
| Пиелонефрит, интерстициальный нефролитиаз, аминоацидурия, ХПН.
| Аминоацидурия, интерстициаль-ный нефрит, ХПН
| Те же заболевания что и при глициринурии, синдром де Тони-Дебре-Фанкони
|
| Лечение
| Режим: активный, двигательный.
Диета: с ограничением белков, количество метионина ограничить до 0,7г/сут (творог, сыр, молочные продукты, мясо, рыба, яйцо, бобовые) на 3-4 нед. затем диету расширить; не рекомендуется - творог, рыбу, яйца.
Увеличение питьевого режима, чтобы диурез был - 3-4 л/сут. прием 400-600 мл. жидкости перед сном, поддержание рН мочи на уровне 7,5 (использование ощелоч. средств. Д-пенициламин назначают с большой осторожностью.
| Симптоматичес-кое, так как принципы терапии не разработаны
| При отсутствии клинических признаков в лечении не нуждается.
При наличии клинических выраженных форм – лечение симптоматическое, так как принципы терапии не разработаны
|
Цистинурия. Заболевание генетически детерминировано, передается по аутосомно-рецессивному типу. Частота 1:7000 новорожденных. Уролитиаз при цистинурии чаще встречается у лиц мужского пола, что связано с анатомическими особенностями строения мочевой системы. Цистиновые камни в почках обнаруживаются примерно у 2% взрослых и у 10% детей, больных мочекаменной болезнью.
Патогенез. При цистинурии имеет место дефект апикального транспорта как основных аминокислот (лизина, аргинина, орнитина), так и цистина, не только в почках, но и в кишечнике. Установлено существование двух транспортных систем (I и II) для цистина и двух (II и III) для диаминомонокарбоновых аминокислот. При этом вторая транспортная система является общей для цистина и диаминомонокарбоновых аминокислот. Она локализуется в люминальной (обращенной в просвет канальца) стенке тубулярного эпителия. Именно генная мутация, приводящая к нарушению работы второго транспортного канала, обусловливает развитие типичного симптомокомплекса цистинурии.
Различают три клинико-генетических варианта (типа) на основании биоптатов слизистой кишечника гомозигот и экскреции с мочой цистина у гетерозигот. I тип болезни встречается наиболее часто, он обусловлен мутациями гена SLC3A1, который локализован на хромосоме 2р16.3. У больных отсутствует тонкокишечный транспорт аминокислот, в том числе и цистина и обусловлен дефектом гена rBAT - транспортного белка. Риск нефролитиаза очень высок. Данный тип цистинурии наследуется аутосомно-рецессивно. Гетерозиготные носители болезни (в том числе родители больного ребенка) экскретируют с мочой нормальные количества цистина (0-100 ммоль на 1 г креатинина). II и III типы цистинурии обусловлены разными аллельными мутациями одного гена SLC7A9, который расположен на хромосоме 19q13.1 и кодирует легкую субъединицу того же транспортного белка. При II типе транспорт цистина в почках снижен наполовину, транспорт других аминокислот в почках и кишечнике отсутствует. Цистинурия III типа, при которой тонкокишечный транспорт указанных аминокислот нормален, обусловлен мутацией в различных локусах.
Морфология. В почечной ткани больных выявляются кристаллы цистина, отмечается поражение тубулярного эпителия, интерстициальные изменения. У части детей наблюдаются признаки интерстициального нефрита, пиелонефрита, нефросклероза, нефролитиаза.
Клиника. Клиническая симптоматика у гомозигот и гетерозигот различна. У гомозигот заболевание может проявляться уже на первом году жизни в виде отставания в физическом развитии, связанного с постоянной потерей таких незаменимых аминокислот, как аргинин и лизин, и признаками инфекции мочевой системы. У детей наблюдаются нарушение мочеиспускания, интоксикация, повышение температуры. В моче часто определяются гематурия, лейкоцитурия и протеинурия. Уролитиаз, нередко с характерными приступами почечной колики, обычно формируется к 3-5 годам, но может выявляться и раньше. У гетерозигот, особенно у девочек, заболевание вначале протекает бессимптомно; его можно заподозрить при обнаружении кристаллов цистина в моче, нередко выявляется микрогематурия. Интерстициальный нефрит формируется медленно и обычно диагностируется в подростковом возрасте или у взрослых.
При всех формах цистинурии наблюдается повышенное выделение цистина с мочой, что приводит у части детей к развитию нефроуролитиаза за счет образования цистиновых камней. В физиологических условиях цистин фильтруется в клубочковом аппарате почек и почти 90% его реабсорбируется в почечных канальцах благодаря деятельности активной транспортной системы. Выведение цистина с мочой у больных цистинурией в 2-4 раза больше, чем в норме, и составляет 250 мг на 1 г креатинина. Нормальной считается экскреция цистина с мочой менее 20 мг/сут. Цистин является малорастворимой аминокислотой (концентрация насыщенного раствора цистина при рН 7,0 составляет не более 400 мг/л), и поэтому при высокой концентрации цистина в моче, особенно в условиях кислой реакции мочи, легко образуются кристаллы, формирующие в последующем цистиновые камни. При цистинурии у пациентов отмечаются симптомы, характерные для нефролитиаза. Для цистинурии характерно наличие повторных, множественных камней, с формированием «коралловидных» камней.
Критерии диагностики цистинурии:
· повышенное выделение цистина с мочой – 250 мг на 1 г Cr (в норме менее 20 мг/сут) определяется в реакции нитропроуссидом натрия или методом хроматографии;
· кислая реакция – высокая концентрация цистина;
· для цистинурии характерно наличие повторных, множественных камней – рентген-контрастных;
· конкременты цистина могут формировать «коралловидные» камни;
· исследование состава камня методами инфракрасной спектроскопии или рентген-дифракции;
· морфология почки: признаки тубуло-интерстициального поражения, отложение цистина (биопсия обычно не проводится);
· генная диагностика.
Лечение. Лечение должно быть направлено на снижение количества нерастворимого цистина в организме. Оно проводится в трех направлениях:
· путем назначения диеты, не содержащей цистина и его предшественников;
· увеличением количества потребляемой жидкости с целью снижения концентрации цистина в моче;
· назначением препаратов переводящих цистин в растворимые формы, повышение рН мочи.
Важным является ощелачивание мочи с целью предупреждения кристаллизации цистина, для чего используются щелочные минеральные воды (до pH>7,5), натрия гидрокарбонат, блемарен, уролит, цитратные смеси. Необходимо учесть, что очень трудно достичь рН >7,5. В последние годы у детей все чаще стали применять локальную литотрипсию. Необходимо отметить, что при всех видах терапии мочекаменной болезни (МКБ) при цистинурии возможно рецидивирование цистиновых камней, что требует постоянного проведения профилактических мероприятий (диетотерапия, питьевой режим, профилактическое применение препаратов). При выявлении у детей признаков пиелонефрита или интерстициального процесса назначается соответствующая терапия.
Прогноз. У детей с цистинурией, особенно осложненной уролитиазом, нередко развивается хроническая почечная недостаточность, связанная с прогрессированием интерстициального процесса.
Профилактика цистинурии базируется на результатах медико-генетического консультирования и определения степени генетического риска повторного рождения больного ребенка.
Для раннего выявления заболевания рекомендуется массовое обследование детей на цистин в моче. При раннем выявлении цистинурии в доклинической стадии рекомендуется диета и наблюдение за детьми в связи с возможностью рецидивирования цистиновых камней и формирования ХПН.
Задание 1
Девочка 6 лет из семьи с отягощенным наследственным анамнезом по патологии мочевыделительной системы в виде МКБ по обеим линиям. Кроме того, у брата 11 месячного возраста также выявлена МКБ. Девочка от первой беременности, протекавшей благоприятно, первых физиологических родов. Масса при рождении 4000 г, длина тела 55 см. Находилась на грудном вскармливании до 1 года. Развивалась удовлетворительно. В 1 год 10 мес. на фоне полного здоровья появились болезненные ощущения внизу живота. При обследовании в лоханке левой почки были выявлены конкременты. В анализах мочи определялись лейкоцитурия, микрогематурия. Была диагностирована МКБ, произведена операция - пиелолитотомия слева. В динамике через 3 года неоднократно девочка лечилась консервативно по поводу двустороннего уролитиаза (рецидив слева), рецидивирующего пиелонефрита. В возрасте 5 лет при обследовании выявлены коралловидные камни почек (рецидив слева). Хронический калькулезный пиелонефрит, рецидивирующее течение.
При поступлении состояние средней тяжести по заболеванию. Жалобы на периодические (1 раз в 2-3 мес.) боли при мочеиспускании. При осмотре: живот при пальпации мягкий, безболезненный во всех отделах. Печень выступала на 0,5 см из-под края реберной дуги. В крови выявлена легкая анемия (Hв 117 г/л), лейкоцитоз до 23 тыс., нейтрофилез (68,4%). Отмечалось повышение в крови уровня IgG (13,3 г/л) и IgE (95 МЕ). Биохимические показатели в пределах нормы. В анализе мочи выявлена лейкоцитурия (18 лейкоцитов в п/зр), протеинурия неселективного характера (0,657 г/сут). Экскреция с мочой уратов и оксалатов соответствовала нормальным значениям, при снижении экскреции калия, натрия, хлора и показателей ацидоаммониогенеза. По данным УЗИ отмечалось увеличение размеров почек, без повышения эхогенности их паренхимы; наблюдалось расширение всех групп чашечек - справа от 0,6 до 1,6 см, слева - от 1,0 до 1,7 см. Были выявлены конкременты: в лоханке правой почки размером 1,8 см, в нижней группе чашечек - 1,4 см; в лоханке левой почки - 1,5 см, в нижней группе чашечек - 1,2 см. При рентгенографическом исследовании органов грудной клетки патологических изменений со стороны легких, сердца и его сосудов обнаружено не было. На экскреторных урограммах выявлялся двусторонний гидронефроз в результате обструкции лоханок конкрементами, формирующийся коралловидный камень.
Вопросы:
1. Каков предположительный диагноз?
2. Какие исследования необходимо провести для уточнения диагноза?
3. Лечение и прогноз заболевания.
Ответы:
1. С учетом клинико-анамнестических данных: рецидивирующее камнеобразование у ребенка, наличие заболевания у сибса и других членов семей - была заподозрена цистинурия.
2. В связи с этим был исследован уровень диаминомонокарбоновых аминокислот в моче. Выявлено многократное увеличение уровня цистина - 598,3 мг/л (норма до 5-20 мг/л), аргинина - 958,4 мг/л (норма до 7-20 мг/л), орнитина - 269,2 мг/л (норма до 3-10 мг/л), лизина - 936,1 мг/л (норма до 2-10 мг/л). Данные изменения являются специфическими для цистинурии. На основании проведенного обследования ребенку был поставлен клинический диагноз: цистинурия (предположительно II типа), рецидивирующая МКБ с формированием двустороннего гидронефроза, рецидивирующий пиелонефрит, активная фаза, ХБП.
3. В качестве терапии: ограничение употребления в пищу белковых продуктов, богатых серосодержащими аминокислотами, обильное ощелачивающее питье. Учитывая формирование крупных конкрементов с нарушением уродинамики и формированием гидронефроза рекомендована литотрипсия. В последующем наряду с диетотерапией, гидратацией и ощелачиванием мочи с лечебной и профилактической целью длительно применялся цистеамин 300-400 мг 3 раза в день.
Таким образом, представлен семейный случай цистинурии, который характеризовался ранней манифестацией заболевания и ранним камнеобразованием.
Обращает на себя внимание наличие достаточно выраженной неселективной протеинурии, что указывает на развитие тубулоинтерстициального процесса, что значительно ухудшает прогноз заболевания. Лечение детей с цистинурией, сопровождающееся формированием больших камней, комбинированное с применением литотрипсии или нефролитостомии с последующим профилактическим лечением, предотвращающим развитие новых камней. К сожалению, поздняя диагностика цистинурии в данном случае потребовала проведения литотрипсии до назначения патогенетической терапии. Прогноз заболевания у больных зависит от эффективности профилактических мероприятий, предупреждающих формирование цистиновых камней и прогрессирование тубулоинтерстициального процесса, а также своевременного лечения рецидивов пиелонефрита.
Лизинурия (аутосомно-рецессивный тип, распространенность - 1:60 000-1:80 000), при которой также нарушается функция транспортера для основных аминокислот, но всасывание цистина практически не нарушено, может клинически не проявляться, но, в манифестных случаях, характеризуется задержкой умственного и физического развития.
Лизинурическое нарушение толерантности к белку обусловлено мутацией базолатерального транспортера (mCAT), который присутствует как проксимальных канальцах, так и в энтероцитах.
3.2.2 В группу заболеваний, связанных с нарушением транспорта нейтральных аминокислот, входят синдром Хартнупа, метионинурия, иминоглицинурия, индиканурия и глицинурия. При болезни Хартнупа, которая передается по аутосомно-рецессивному типу (распространенность – 1:26 000–1:200 000) в кишечнике и почках нарушается транспорт 12 нейтральных аминокислот, что приводит к генерализованной аминоацидурии. При этом уровень всех аминокислот в крови остается в пределах нормы, за исключением триптофана. Невсосавшийся в кишечнике триптофан, подвергается бактериальному разложению с образованием индола и индоксила, которые всасываются, подвергаются в печени детоксикации и в виде метаболитов экскретируются с мочой.
Основным клиническим проявлением болезни является фотосенсибилизация кожных покровов, а также может наблюдаться мозжечковая атаксия, поражение пирамидных путей, возможны немотивированные подъемы температуры тела и незначительная умственная отсталость. Были клонированы и функционально охарактеризованы несколько транспортеров нейтральных аминокислот: транспортер системы А (семейство СГЛТ), транспортер системы ASC (семейство глютамат/нейротрансмиттера). При болезни Хартнупа развивается пеллаграподобный синдром, и хотя обычно он требует лечения, в отдельных случаях возможно пероральное применение никотинамида, неомицина или триптофана, высокобелковой диеты.
Метионинурия обусловлена изолированным дефектом транспорта метионина нейтральной кислоты, транспорт которой не нарушен при синдроме Хартнупа. Аутосомно-рецессивное наследование, возможно развитие тетании, задержки умственного развития.
Семейная иминоглицинурия - аутосомно-рецессивное состояние (распространенность - 1:15 000), при котором нарушены кишечное всасывание и канальцевая реабсорбция пролина, гидроксипролина и глицина, при отсутствии клинических проявлений. Конечным метаболитом всосавшегося из кишечника индола является образующийся в печени индикан. Индиканурия характерна для многих состояний, связанных со стазом в кишечнике, а также для фенилкетонурии, но максимально выражена при врожденных метаболических дефектах обмена триптофана. Уже в моче индикан может образовывать синий индиго, придающий соответствующий цвет моче и детским пеленкам. Семейное заболевание, именуемое «синдромом голубых пеленок», характеризуется мальабсорбцией триптофана, гиперкальциурией и нефрокальцинозом с последующей почечной недостаточностью.
Глицинурия обусловлена дефектом глицина, и иногда ассоциирована с глюкозурией (так называя глюкозоглицинурия). Считается, что и иминоглицинурия и глицинурия обусловлены мутациями натрий- и хлорзависимых транспортеров аминокислот, которые принадлежат семейству GABA/нейротрансмиттеров, куда входят также транспортер норэпинефрина, дофамина, серотонина, холина, таурина, бетаина, L-глицина и пролина.
Ренальный тубулярный ацидоз (РТА)
Одной из основных функций почек является поддержание кислотно-щелочного равновесия, которое осуществляется путем секреции ионов Н+.
Секреция ионов Н+ происходит в проксимальных и дистальных канальцах.
Регуляция кислотно-основного равновесия. Почки принимают участие в поддержании кислотно-основного равновесия крови, экскретируя кислые продукты обмена. В процессе метаболизма в организме образуется некоторые количество кислот, часть ионов Н+ поступает с пищей, в связи, с чем возникает необходимость выведения избытка кислот из организма.
Нарушение реабсорбции бикарбоната в проксимальных канальцах является дефектом, обусловленным дефицитом карбоангидразы II, лежащим в основе проксимального ренального тубулярного ацидоза или ацидоза II типа.
Дефицит карбоангидразы II является аутосомно-рецессивным расстройством, и характеризуются ренальным тубулярным ацидозом (РТА), с проксимальным и дистальным компонентом, остеопорозом и кальцификацией. Карбоангидраза катализирует обратимую гидратацию диоксида углерода (CO2), и тем самым ускоряет превращение двуокиси углерода и воды в ионы водорода (H+) и бикарбонат (HCO3-). Важным местом реабсорбции НСО3- является проксимальный каналец, где 90% профильтровавшегося НСО3- всасывается обратно. В проксимальных канальцах реабсорбция бикарбоната осуществляется комбинированным действием люминальной карбоангидразы IV типа (CA4) и цитозольной карбоангидразой II типа (СА2), люминальным обменником. Это осуществляется не за счет прямого транспорта, а посредством специального механизма, в котором участвует карбоангидраза и Na+/H+- обменник. (рис. 5).
Таким образом, интенсивная секреция Н+ используется для возвращения профильтровавшихся бикарбонатов. На данном этапе ионы Н+ находятся в постоянном круговороте, обеспечиваемом карбоангидразой, и элиминаций Н+ с мочой не происходит. Ионы водорода секретируются на всем протяжении канальцев. Далее в дистальных участках нефрона экскреция кислот осуществляется двумя механизмами: за счет связывания Н+ с фосфатами и с аммиаком (NH3). Ближе к собирательным трубочкам к Na+/H+ обменнику подключается (а затем превалирует) Н+-АТФ-аза. Эти процессы происходят во вставочных клетках собирательных трубок.

Cl - ионы хлора; H2CO3- - углекислота; К+ - ионы калия; Na+ - ионы натрия
Рисунок 5 - Дефицит карбоангидразы II
Нарушение реабсорбции бикарбоната в проксимальных канальцах является основным дефектом II типа или проксимального РTA. В дистальном нефроне карбоангидразы II типа экспрессируется в интерколатаральных клетках кортикальных собирательных трубочек. Там карбоангидраза II типа играет важную роль в катализировании конденсации гидроксильных ионов, генерируемых протон-транслокационных Н+ - аденозинтрифосфатазы (H+АТФазы), с диоксидом углерода с образованием бикарбоната. Дефицит карбоангидразы II типа, увеличивает внутриклеточный рН с ухудшением деятельности протон- транслокационной Н-АТФазы. Ингибиторы карбоангидразы являются слабыми диуретиками, которые блокируют реабсорбцию бикарбоната благодаря своему воздействию на карбоангидразу.
Задание 2
Если взаимодействие рСО2 и Н2О приводит к образованию одной молекулы НСО3- и Н+, почему тогда удвоение величины рСО2 в плазме крови (с 40 до 80 мм рт. ст.) приводит к снижению значения рН плазмы?
Ответ
Повышение рСО2 до 80 мм рт. ст. увеличивает Н+ с 40 до 80 ммоль/л. Нормальное значение НСО3- равно 24 ммоль/л. Поэтому в норме Н+ = 24 (40 мм рт. ст.): (24 ммоль/л) = 40 нМ; но при задержке в организме СО2 (рСО2), Н+=24 (80 мм рт. ст.): (24,00004 ммоль/л)=80 нМ. То есть концентрация НСО3 в плазме становится намного выше, чем концентрация Н+. Поэтому изменение рСО2 приводит к пропорционально большему изменению Н+ по сравнению с НСО3-.
Дистальный РТА развивается вследствие недостатка карбоангидразы во вставочных клетках КСТ. Собирательный отдел является основным местом подкисления мочи, где происходит окончательная реабсорция от 5% до 10% отфильтрованного бикарбоната и ионов водорода (H+). Дистальный нефрон состоит из нескольких отдельных сегментов, например, соединительной трубочки, корковых собирательных канальцев и канальцев мозгового вещества. Собирательные трубки содержат два типа клеток: основные, которые транспортируют натрий, калий и воду; и вставочные, которые секретируют ионы водорода и бикарбонат (НСО3-). Нарушение подкисления в дистальном отделе нефрона может привести дРТА. Различные типы ренального тубулярного ацидоза представлены в таблице 6 и 7.
Таблица 6 - Различные типы ренального тубулярного ацидоза
| | РТА I типа,
дРТА
| РТА II типа,
пРТА
| РТА IV типа Гиперкалие-мический
|
| HCO3-
| Может быть очень низким
| Вариабельно
низкий
| Обычно 15-18 ммоль/л
|
| K+
| Низкий
| Относительно низкий
| Высокий
|
| рН мочи
| >6,0 (полный дРТА)
| <5,5 (если НСО3-) достаточно низкий
| Обычно <5,5
|
| U-B pCO2
| Снижен
| Нормальный
| Снижен
|
| FE HCO3
| <5%
| >15% низкий
| <5,5
|
| Нефрокальциноз/
литиаз
| Да
| Нет
| Нет
|
| U-B pCO2 – разница между рСО2 мочи (U) и крови (В)
FE – фракционная экскреция
|
Клинические признаки
Наиболее частые признаки РТА:
· задержка развития
· рвота, снижение аппетита
· различной степени мышечная слабость (табл. 7).
| Клиника
| Интерпретация
лабораторно-инструментальных
данных
| Дифференциальный
диагноз
| Лечение
|
| Отставание в росте. Мышечная слабость, вплоть до параличей, поли-дипсия, полиурия.
Рахитоподобная деформация скелета. Кризы обезвоживания.
В грудном возрасте недостаточная прибавка массы тела, анорексия, иногда рвота, запоры.
| Полиурия ( N суточный диурез составляет 60-80% от выпитой жидкости).
Гипо-, изостенурия (N уд.вес в зависимости от диеты 1010-1025).
Щелочная реакция мочи (рН мочи не ниже 6,8).
Дефицит оснований в крови (в N ВЕ = (+2) – (-5).
Дизэлектролитемия:
Гиперхлоремия (N=90-100ммоль/л), гипокалиемия (N= 3,5-5,5ммоль/л),
гипокальцемия (N=2,2-2.9ммоль/л), гипофосфатемия (N= 1,2-2,2ммоль/л).
Ro-исследование:
Остеомаляция, нефрокаль-циноз.
| Фосфат диабет:
Снижен только уровень Р в крови. Не отмечается измене-ний в составе электролитов, КОС, аминокислот крови.
Изменения мочи не характерны.
Болезнь де-Тони-Дебре-Фанкони:
Метаболический ацидоз сочетается с гиперамино-ацидурией, фосфатурией, глюкозурией.
Рахит: характеризуется отсутствием изменений со стороны мочи.
КОС изменяется незначительно. Электролитный состав крови почти не меняется, за исключением содержания Са, редко Р.
Заболевание начинается до 1года.
| 1.Введение гидрокарбоната натрия из расчета: ВЕ (дефицит гидрокарбонатов) х 1/3 массы тела 5% р-ра (в100мл содержится 60 ммоль гидрокарбоната).
2. Коррекция диз-электролитемии: препараты К+, Са+.
3. Витамин Д 50000 МЕ/сут
4.Гипотиазид 2-3мг/кг длительно (1-5лет).
|
Таблица 7 – Ренальный тубулярный ацидоз
РТА II типа (проксимальный РТА, пРТА)
Характеризуется гиперхлоремическим метаболическим ацидозом с нормальной или немного сниженной сывороточной концентрацией калия и сохраненной способностью снижать рН мочи ниже 5,5. При пРТА нарушенная реабсорбция HCO3- в проксимальном канальце компенсируется реабсорбцией в дистальном канальце при уровне бикарбонатов в плазме не выше 18 ммоль/л. При более высоком уровне HCO3- рН мочи не снижается, и у больных продолжается потеря натрия с мочой, с развитием вторичного гиперальдостеронизма и умеренной гипокалиемии. Почечная потеря калия возникает за счет повышения альдостерона в ответ на повышение доставки Na в дистальный каналец. При снижении рН мочи ниже 5,5 - HCO3- плазмы снижаются ниже пороговых значений, что ведет к дистальной секреции Н+. Эта способность продуцировать кислую мочу, объясняет отсутствие рахита, гиперкальци-урии и нефрокальциноза при пРТА. пРТА может наблюдаться как изолированный дефект или, почти всегда у детей, как часть генерализованного проксимального канальцевого нарушения с почечным синдромом Фанкони.
Причины РТА II типа
ü Первичный пРТА: транзиторный (с ранним началом), персистирующий (начало во взрослом возрасте), или наследственный (дефицит карбоангидразы).
ü Вторичный пРТА: наследственный синдром Фанкони, гиперпаратиреоидизм, витамин D-зависимость и дефицит, синдром Lowe, митохондриальные цитопатии.
Клиника пРТА II типа - первые симптомы заболевания появляются в возрасте 3-18 мес., чаще у мальчиков.
· Синдром дистрофии и обменных нарушений: снижение аппетита, тошнота, рвота, эпизоды дегидратации, замедление темпов нарастания массы тела, отставание в росте, артериальная гипотензия, «беспричинные» подъемы температуры
· Костные изменения выражены слабо, наблюдаются искривления большеберцовых и бедренных костей
· Лабораторно: в анализах мочи: щелочная реакция, повышенное содержание бикарбонатов; в анализе крови: гипокалиемия, гиперхлоремия, метаболический ацидоз.
Лечение
· Коррекция ацидоза (бикарбонат натрия в дозе 1-3 ммоль/кг у детей старше 5 лет, 4-15 ммоль/л – у детей младшего возраста).
· Коррекция электролитных нарушений (гипокалиемии, гипокальциемии): хлорид калия, карбонат кальция.
· Профилактика прогрессирования нефрокальциноза (цитраты в виде смесей: уралита, блемарена, смеси Олбрайта).
· Лечение остеопатии активными метаболитами витамина D3: (альфакальцидол 0,5-1 мкг/сут). Осторожно ® может усилить гиперкальциурию.
РТА I типа (классический дистальный РТА, дРТА)
дРТА характеризуется гиперхлоремическим метаболическим ацидозом, сопровождающимся снижением экскреции кислых валентностей (Н+) в дистальных канальцах. При дРТА почка не способна подкислять мочу при любом уровне ацидоза. Поэтому у этих больных может развиться тяжелый метаболический ацидоз, угрожающий жизни. Он сопровождается гипокалиемией, потерей натрия, вторичным гиперальдостеронизмом и нефрокальцинозом.
Формы заболевания: изолированные первичные нарушения при спорадических, или наследственных (аутосомно-доминантных или рецессивных, иногда сочетающихся с нейросенсорной тугоухостью) заболеваниях.
Первичный дефект поражает апикальную Н+-АТФазу (рецессивный тип с тугоухостью), базолатеральную Н+/К+-АТФазу (рецессивный тип) или базолатеральный НСО3- /Cl- обменник (при доминантном типе). При всех трех формах общей является неспособность снижать рН мочи ниже 6,0 и присутствии спонтанного ацидоза (полный дРТА). Вторичный дРТА может развиться на фоне течения таких болезней почек, как нефрокальциноз, аутоиммунные болезни или при воздействии токсинов или препаратов, поражающих дистальный каналец или собирательные канальцы (табл. 8).
Таблица 8 – Виды дистального РТА
| Виды дистального ренального тубулярного ацидоза (I типа)
|
| I первичный
ü наследственный (аутосомно-рецес-сивный с- или без тугоухости, аутосомно-доминантный)
ü спорадический
ü неполный дистальный РТА
II вторичный
• Наследственные заболевания:
ü синдром Элерса-Данлоса
ü синдром Марфана
ü наследственный эллиптоцитоз
ü медуллярная кистозная болезнь
ü серповидноклеточная анемия
ü остеопороз
• Аутоиммунные заболевания:
ü гипергаммаглобулинемия
ü синдром Шегрена, СКВ
ü хронический активный гепатит
| • Гиперкальциурия и нефрокальциноз
ü первичный гиперпаратиреоидизм
ü интоксикация витамином D
ü идиопатическая гиперкальциурия
ü гипертиреоидизм
• Тубулоинтерстициальные
заболевания
ü обструктивная уропатия
ü медуллярная губчатая почка
ü отторжение почечного трансплантата
ü острая почечная недостаточность
• Лекарства и токсины
ü анальгетическая нефропатия
ü амфотерицин В, литий, цикламат, ртуть
• Гипонатрийурические состояния
ü нефротический синдром
ü цирроз печени
|
Схема патогенеза дРТА представлена на рис. 6
 Рисунок 6 – Схема патогенеза дРТА (8)
Рисунок 6 – Схема патогенеза дРТА (8)
Клиника дРТА у детей: Манифестация в возрасте 3 мес-2 лет. При инфантильном типе (синдром Лайтвуда) болезнь начинается в грудном возрасте, при синдроме Батлера-Олбрайта – не ранее 2-3 лет. Чаще болеют девочки.
· Синдром дистрофии и обменных нарушений: снижение аппетита, тошнота, рвота; полиурия, полидипсия, дегидратационные кризы, сочетающиеся с расстройством кровообращения, аритмиями сердца, сонливостью, вялыми параличами; задержка роста и прибавки массы тела, субфебрилитет;
· Костные изменения: выраженная вальгусная деформация костей нижних конечностей, патологические переломы, боли в костях у детей более старшего возраста.
· Лабораторно: в анализах мочи – щелочная реакция (рН не ниже 6,0), умеренная протеинурия, микрогематурия и лейкоцитурия; в анализе крови – гипокалиемия, гиперкальциурия, гипонатриемия, гиперхлоремия, декомпенсированный метаболический ацидоз.
Рентгенография костей – Х-образное искривление нижних конечностей, остеопороз, патологические переломы.
Рентгенография мочевыводящих путей – нефрокальциноз, нефролитиаз (рис.7).
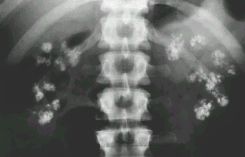
Рисунок 7 - Нефрокальцино