Представление о механизмах реакций. Гомо- и гетеролитический разрыв связей. Представление о промежуточных частицах: радикалы, карбокатионы, карбанионы. Классификация реагентов: радикалы, нуклеофилы, электрофилы.
Механизм реакции — это детальное описание процесса превращения реагентов в продукты, включ-е в себя как можно более полное описание состава, строения, геометрии, энергии и других св-тв интермедиатов, переходных состояний и продуктов.
Гомолитический разрыв связи — разрыв, когда каждому атому отходит по одному электрону. Характерен для обменного механизма образования ковалентной связи.
Гетеролитический разрыв связи — разрыв, когда в рез-те образуются положительно и отрицательно заряж-е частицы, т.к. оба электрона из общей электронной пары остаются при одном из атомов. Характерен для донорно-акцепторного механизма образования ковалентной связи.
Карбкатион — частица, в кот. на атоме углерода сосредоточен положит-й заряд, атом углерода имеет вакантную p-орбиталь. Карбкатион — сильная кислота Льюиса, обладает электрофильной активностью. Химич. св-ва:
· Взаимодействие с нуклеофилами.
· Способность к β-элиминированию — отщеплению протона с образованием кратной связи.
· Перегруппировка в более стабильный карбкатион — изомеризация первичного в более стабильный вторичный или третичный карбкатион.
Карбанион — анион, сод-й чётное число электронов со свободной электронной парой на четырехвалентном атоме углерода. К карбанионам относят как анионы с локализованным на углеродном атоме отрицательном заряде, так и анионы с делокализованным отрицательным зарядом, у которых по крайней мере в одной из канонических структур заряд локализован на атоме углерода. Хим. св-ва:
· Взаимодействие с электрофилами.
· Окисление до радикалов.
Свободные радикалы — частицы (как правило, неустойчивые), сод-е один или неск. неспаренных электронов на внешней электрон. оболочке. Радикал может образ-ся в рез-те потери одного электрона нерадикальной молекулой или при получении одного электрона нерадикальной молекулой.
Кислоты и основания (Бренстед, Льюис)
Протолитическая (протонная) теория кислот и оснований Брёнстеда — Лоури (1923). Согласно этой теории кислотами явл-ся молекулы или ионы, способные быть в данной реакции донорами протонов, а основаниями явл-ся молекулы или ионы, присоединяющие протоны (акцепторы). Кислоты и основания получили общее название протолитов.
Сущностью кислотно-основного взаимод-я явл. передача протона от кислоты к основанию. При этом кислота, передав протон основанию, сама становится основанием, т.к. может снова присоединять протон, а основание, образуя протонированную частицу, становится кислотой. Таким образом, в любом кислотно-основном взаимодействии участвуют две пары кислот и оснований, названные Бренстедом сопряженными.
Электронная теория Льюиса. В теории Льюиса (1923 г.) на основе электронных представлений было ещё более расширено понятие кислоты и основания. Кислота Льюиса — молекула или ион, имеющие вакантные электронные орбитали, вследствие чего они способны принимать электронные пары. Это, например, ионы H — протоны, ионы металлов (Ag+, Fe3+), оксиды некоторых неметаллов (например, SO3, SiO2), ряд солей (AlCl3), а также такие вещ-ва как BF3, Al2O3. Кислоты Льюиса, не содержащие ионов водорода, называются апротонными. Протонные кислоты рассматриваются как частный случай класса кислот. Основание Льюиса — это молекула или ион, способные быть донором электронных пар: все анионы, аммиак и амины, вода, спирты, галогены.
Пути использования алканов
Алканы находят широкое применение во многих сферах деят-ти человека. Ни один из нас уже не может представить жизнь без природного газа, основой кот. является метан. Из него также производят технический углерод (сажу), кот. используется в производстве шин, типографской краски. Соединения алканов прим-ся в качестве хладагентов в домашних холодильниках. Ацетилен, кот. получают из метана, используется для сварки и резки металлов. Среди соединений алканов можно выделить галогенопроизводные, такие как хлороформ, четырёххлористый углерод, являющиеся одними из лучших растворителей. Алканы могут применяться в качестве моторного топлива (метан, пропан, бутан), кот. мало загрязняет окружающую среду. Вазелиновое масло (смесь жидких углеводоpодов с числом атомов углерода до 15) - пpозpачная жидкость без запаха и вкуса, используется в медицине, паpфюмеpии и косметике.Вазелин (смесь жидких и твеpдых пpедельных углеводоpодов с числом углеpодных атомов до 25) пpименяется для пpиготовления мазей, используемых в медицине.Паpафин (смесь твеpдых алканов С19-С35) - белая твеpдая масса без запаха и вкуса (tпл= 50-70°C) - пpименяется для изготовления свечей, пpопитки спичек и упаковочной бумаги, для тепловых пpоцедуp в медицине и т.д.
Sp-гибридизация
Происходит при смешивании одной s- и одной p-орбиталей. Образуются две равноценные sp-атомные орбитали, расположенные линейно под углом 180 градусов и направленные в разные стороны от ядра центрального атома. Две оставшиеся негибридные p-орбитали располагаются во взаимно перпендикулярных плоскостях и участвуют в образовании π-связей, либо занимаются неподелёнными парами электронов.
Sp2-гибридизация
Происходит при смешивании одной s- и двух p-орбиталей. Образуются три гибридные орбитали с осями, расположенными в одной плоскости и направленными к вершинам треугольника под углом 120 градусов. Негибридная p-атомная орбиталь перпендикулярна плоскости и, как правило, участвует в образовании π-связей
26. Алкины. Восстановление тройной связи до двойной: каталитическое гидрирование и восстановление натрием в жидком аммиаке, использование в синтезе (Z)- и (Е)- алкенов.
Тройная связь представляет собой одну s -связь С-С и две p -связи. При переходе от двойной к тройной связи средняя энергия p -связи снижается. Это означает, что тройная связь менее стабильна, чем двойная. Сам ацетилен неустойчивое соединение и способен к спонтанному взрывному распаду на элементы. Молекула ацетилена имеет линейное строение, что обусловлено sp-состоянием атомов углерода. Тройная связь в алкинах характеризуется более высокой поляризуемостью, чем в алкенах RCº C = 5,96; RC=C=4,17.
Метод каталитического гидрирования, наряду с другими важнейшими процессами органической химии, широко используется в настоящее время в. Внедрение гидрирования в технику явилось стимулом для широкого развития процессов облагораживания топлива, синтезов, из окислов углерода и многочисленных реакций восстановления. Гидрирование алкинов происходит примерно в тех же условиях и в присутствии тех же катализаторов, что и гидрирование алкенов. Первая стадия гидрирования ацетилена до этилена более экзотермична, чем вторая, где этилен превращается в этан: 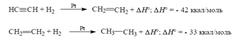
Из этих данных следует, что гидрирование алкинов, в принципе, можно остановить на стадии образования алкена. Однако с большинством катализаторов алкины гидрируются прямо до алканов: 
Восстановление алкинов натрием или литием в жидком аммиаке или в аминах дает транс -алкены:

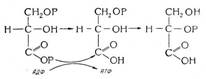
Алке́ны (олефины, этиленовые углеводороды) — ациклические непредельные углеводороды, содержащие одну двойную связь между атомами углерода, образующие гомологический ряд с общей формулой CnH2n. Атомы углерода при двойной связи находятся в состоянии sp² гибридизации, и имеют валентный угол 120 °C. Простейшим алкеном является этилен (C2H4). По номенклатуре названия алкенов образуются от названий соответствующих алканов заменой суффикса «-ан» на «-ен»; положение двойной связи указывается арабской цифрой. Е-Изомеры – это такие геометрические изомеры, в которых старшие заместители у углеродных атомов двойной связи находятся по разные стороны относительно двойной связи. Z- Изомеры это такие геометрические изомеры, в которых старшие заместители у углеродных атомов двойной связи находятся по одну сторону относительно двойной связи (от немецкого слова «zusamen» - вместе). Обозначение Е- и Z- ставят перед названием соединения по номенклатуре ИЮПАК и заключают в скобки (обозначение цис - и транс- в скобки не заключается). Например:

Номенклатура аренов
Простейший ароматический углеводород состава C6HбC6Hб имеет тривиальное название бензол. Все остальные углеводороды этого ряда могут быть названы как замещенные производные бензола, или имеют свои тривиальные названия. При этом по традиции, установившейся в русском языке, почти все тривиальные названия гомологов бензола также имеют окончание -ол. Например: C6H5CH3C6H5CH3 — метилбензол, или толуол; C6H4(CH3)2C6H4(CH3)2 — диметилбензол, или ксилол; C6H5CH(CH3)2C6H5CH(CH3)2 — изопропилбензол, или цимол. Как исключение C6H3(CH3)3C6H3(CH3)3 или 1, 3, 5-триметилбензол имеет название мезитилен. По правилам IUPАС все названия ароматических углеводородов характеризуются окончанием -ен. Соответственно: бензен, толуен, ксилен, цимен, стирен и т. д.
На практике для образования названия дву- и более замещенных одноядерных аренов чаще используют следующие варианты:
1. В основу названия положено тривиальное название арена (толуол, стирол и т.д.), для обозначения месторасположения боковых цепей используют русские буквы (о-, м-, п-) или латинские (o-, m-, p-), что означает орто-, мета- или пара-положения бензольного кольца. Алкильные радикалы или функциональные группы называются в соответствии с номенклатурой ИЮПАК: метил-, этил-, изопропил-, амино-, гидроксо-, нитро- и т.д. Часто такие правила используются для образования названий ароматических соединений других классов - аминобензолов, фенолов и др., содержащих разные заместители.

2. Реже используются названия, в основу которых положено слово "бензол", а месторасположение радикалов-заместителей обозначается цифрами. Называя более сложные производные бензола, как и в случае алициклических соединений, из возможных порядков выбирают тот, при котором сумма цифр номеров заместителей будет наименьшей. При этом нет общепринятых правил порядка нумерации атомов бензольного ядра. По Женевской номенклатуре номер 1 присваивают тому атому заместителя, с которым непосредственно связан атом-заместитель с наименьшим атомным весом (например, при наличии в ядре —Сl и —ОН номер 1 получает атом, несущий —ОН, но при наличии —NO2NO2 и —ОН — атом, несущий —NO2; в замещенных производных гомологов бензола начало нумерации определяет простейшая боковая цепь. Для многоядерных аренов правилами номенклатуры IUPAC установлен перечень названий, положенных в основу номенклатуры конденсированных многоядерных карбоциклических систем, правила ориентации их формул и порядок нумерации атомов. В номенклатуре используются тривиальные названия (нафталин, фенантрен, антрацен) с указанием месторасположения заместителей. Например, для производных нафталина можно также использовать оба правила, описанные выше для одноядерных аренов:

Природа связей в молекуле бензола Молекула бензола содержит систему сопряженных связей. Все шесть атомов углерода циклической молекулы бензола С6H6 лежат в одной плоскости. Между атомами углерода в плоскости кольца действуют σ-связи; такие же связи имеются у каждого атома углерода с атомами водорода. На осуществление этих связей атомы углерода затрачивают по три электрона. Облака четвертых валентных электронов атомов углерода, имеющих форму восьмерок, расположены перпендикулярно к плоскости молекулы бензола. Каждое такое облако перекрывается одинаково с электронными облаками соседних атомов углерода. В молекуле бензола образуются не три отдельные π-связи, а единая π-электронная система из шести электронов, общая для всех атомов углерода. Связи между атомами углерода в молекуле бензола совершенно одинаковые. Все связи между атомами углерода в бензоле равноценны, чем и обусловлены характерные свойства бензольного ядра. Наиболее точно это отражает структурная формула бензола в виде правильного шестиугольника с окружностью внутри. (Окружность символизирует равноценность связей между атомами углерода.) Однако часто пользуются и формулой Кекуле с указанием двойных связей.
Кислотно-основные свойства спиртов. Алкоголяты металлов, их основные и нуклеофильные свойства. Реакции нуклеофильного замещения с участием спиртов. Примеры биологически важных реакций нуклеофильного замещения с участием эфиров фосфорных кислот.
спирты являются слабыми ОН-кислотами Бренстеда и жесткими кислотами по Пирсону. По кислотности спирты близки к воде. Кислотные свойства спиртов определяются способностью к протонизации атома водорода гидроксильной группы. Последняя обусловливается не только разницей в электроотрицательностях между атомами кислорода (3,5) и водорода (2,1), но и природой радикала. Метанол (pKa = 15,5), несколько более сильная кислота, чем вода (pKa = 15,7), но большинство спиртов являются более слабыми кислотами, чем вода. Причиной этого являются стерические препятствия, мешающие в разветвленных спиртах сольватации образующегося алкоксид-аниона. Сольватация стабилизирует алкоксид-анион и следовательно усиливает кислотные свойства. Реакции с участием нуклеофильного центра. Высокая электроотрицательность атома кислорода (3,5 по шкале Полинга), являющегося основным центром, позволяет рассматривать спирты как слабые n-основания Бренстеда и жесткие основания по Пирсону.спирты способны образовывать соли оксония только с сильными протонными кислотами и жесткими кислотами по Пирсону (фторид бора, хлорид цинка и др.Таким образом, спирты обладают слабыми кислотными и слабыми основными свойствами, т.е. являются амфипротонными соединениями.При достаточно высокой температуре и в отсутствие хорошего нуклеофила протонированные спирты способны к реакции, т.е. к реакции дегидратации.Будучи жесткими основаниями, вследствие низкой поляризуемости и высокой электроотрицательности, атома кислорода спирты являются слабыми нуклеофилами. Кислоты Бренстеда протонируют атом кислорода гидроксигруппы.
42. Внутри- и межмолекулярная дегидратация спиртов .: Дегидратация спиртов может осуществляется в двух направлениях: внутримолекулярно и межмолекулярно. Направление дегидратации зависит от природы спирта и условий проведения реакции.При внутримолекулярной дегидратации спирта образуется ненасыщенный этиленовый углеводород, в результате межмолекулярной дегидратации – простой эфир. Так, при нагревании спиртов с такими водоотнимающими веществами, как концентрированная H2SO4, H3PO4, безводная щавелевая кислота, оксид алюминия и т.д., образуются ненасыщенные соединения этиленового ряда.Реакционная способность спиртов к дегидратации, то есть к образованию из них этиленовых соединений, изменяется в таком порядке:третичные спирты > вторичные спирты >первичные спирты.Некоторые третичные спирты дегидратируются настолько легко, что их можно перегнать только в случае, если предотвратить попадание в них даже лабораторного воздуха, который содержит в незначительных количествах пары кислот.Дегидратация спиртов в присутствии концентрированной H2SO4 в зависимости от температуры, соотношения объемов спирта и кислоты может осуществляться с образованием разных продуктов. Например, этиловый спирт при 105оС образует с серной кислотой кислый сложный эфир – этилсерную кислоту (реакция 1). При избытке спирта и более высокой температуре (130 –140оС) осуществляется межмолекулярная дегидратация, главным продуктом которой является диэтиловый эфир (простой эфир; реакция 2). При температуре выше 160оС этилсерная кислота разлагается с образованием этилена (реакция 3):

43. Окисление первичных и вторичных спиртов. Спирты при 300-400оС и в присутствии медных и других катализаторов окисляются кислородом воздуха. Такие окислители, как KMnO4, хромовая смесь, окисляют спирты уже при комнатной температуре. В зависимости от того, какой это спирт – первичный, вторичный или третичный – при окислении образуются разные продукты.
Первичные спирты при окислении дают альдегиды с таким же количеством углеродных атомов, как и в молекуле исходного спирта. Альдегиды в этих условиях могут окисляться в карбоновые кислоты. Чтобы избежать дальнейшего окисления, альдегиды необходимо быстро выводить из реакционной смеси
Первичные спирты можно также окислить в альдегиды мелко раздробленной медью. Нагретой до 280-300оС. В этих условиях от молекулы спирта отщепляются два атома водорода и в молекуле органического вещества, которая при этом образуется, появляется двойная связь углерод – кислород (>C=O). Такое превращение спиртов называется дегидрированием:

Вторичные спирты при окислении, а также при дегидрировании, превращаются в кетоны:
Третичные спирты окисляются достаточно трудно с одновременным разрывом углеродной цепи их молекул и образования смеси карбоновых кислот и кетонов. Такое окисление этих спиртов связано с тем, что в условиях реакции окисления они дегидратируются и превращаются в этиленовые углеводороды, которые в присутствии сильного окислителя окисляются с разрывом молекулы по месту двойной С=C – связи
Сульфирование
Сульфирование фенола осуществляют путем нагревания с концентрированной серной кислотой. Температура проведения реакции решающим образом определяет строение образующихся оксибензолсульфокислот. орто -Изомер, скорость образования которого выше, чем пара -изомера, является доминирующим продуктом, если температура проведения реакции не превышает 100 °С. Его называют кинетическим продуктом. Напротив, при более высоких температурах основным продуктом является пара -изомер, скорость образования которого ниже, но он обладает высокой термодинамической устойчивостью Реакция электрофильного ароматического сульфирования обратима и при нагревании орто -оксибензолсульфокислоты, с серной кислотой выше 100 °С получают пара -изомер – продукт термодинамического контроля реакции. Алкилирование. В отличие от алкилирования фенола по гидроксигруппе, которое идет в щелочной среде, введение алкильных заместителей в ароматическое кольцо фенола протекает при действии галогеналканов, спиртов или алкенов в присутствии катализаторов – минеральных кислот или кислот Льюиса (реакция Фриделя-Крафтса). Пикриновая кислота. Наличие в ядре трех нитрогрупп резко увеличивает кислотность фенольной группы. Пикриновая кислота является, в отличие от фенола, уже довольно сильной кислотой. Наличие трех нитрогрупп делает пикриновую кислоту взрывчатой, она используется для приготовления мелинита. Для получения мононитрофенолов необходимо использовать разбавленную азотную кислоту и проводить реакцию при низких температурах:Получается смесь о- и п-нитрофенолов с преобладанием о-изомера. Эта смесь легко разделяется благодаря тому, что только о-изомер обладает летучестью с водяным паром. Большая летучесть о-нитрофенола объясняется образованием внутримолекулярной водородной связи, в то время как в случае.п-нитрофенола возникает межмолекулярная водородная связь.
47. Карбоксилирование фенолятов щелочных металлов. Салициловая кислота. Фенолокарбоновые кислоты получают при взаимодействии фенолятов щелочных металлов с углерода (IV) оксидом.Салициловая (о-гидроксибензойная) кислота является одной из важнейших фенолокарбоновых кислот. Она используется для получения лекарственных препаратов (натрия салицилата, ацетилсалициловой кислоты, фенилсалицилата, метилсалицилата), в синтезе красителей, в производстве душистых веществ (сложных эфиров), для получения кумарина и др.Стадии производства салициловой кислоты:1) получение безводного натрия фенолята:

2) карбоксилирование натрия фенолята углерода (IV) оксидом:

Фенол как побочный продукт отгоняют; 3) разложение сырого натрия салицилата:

Труднорастворимая салициловая кислота выпадает в осадок;
4) отделение и очистка салициловой кислоты.Получаемая техническая салициловая кислота содержит до 99 \% чистого продукта. Салициловая кислота, предназначенная для получения лекарственных веществ, должна быть очищена возгонкой.
48. Окисление фенолов. Повышенная электронная плотность в ядре фенола делает его чувствительным к воздействию окислителей.В зависимости от природы окислителя и условий проведения реакции образуются различные продукты окисления фенола.1) При окислении фенола пероксидом водорода в прис
утствии железного катализатора получается орто-бензохинон через промежуточное образование пирокатехина:
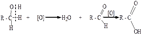
2) Сильные окислители, такие как хромовая смесь (K2Cr2O7 + H2SO4), броматы (KBrO3, H2SO4) окисляют фенол до пара-бензохинона через промежуточное образование гидрохинона:

3) При более энергичном действии окислителей происходит разрушение бензольного ядра.Благодаря склонности к окислению фенолы могут окрашиваться при хранении на воздухе.
49. Хиноны и их биологическая роль. Хинонами называются шестичленные циклические дикетоны с двумя двойными связями.Наибольшее практическое значение из них имеет парахинон, получаемый окислением гидрохинона или анилина. Парахинон – исходный продукт при синтезе гидрохинона. Характерное для хинона расположение двойных связей обусловливает окраску ряда соединений.Нафтохиноны – производные нафталина, содержащие хиноидное ядро. Наибольшее значение имеет 1,4-нафтохинон, который можно получить при окислении нафталина.По ряду своих свойств 1,4-нафтохинон сходен с п-бензохиноном. Он кристаллизуется в виде желтых игл, летуч, обладает острым раздражающим запахом.Ядро 1,4-нафтохинона лежит в основе витамина К, или антигеморрагического витамина (препятствующего появлению кровоизлияний). Витамин К представляет собой 2-метил-3-фитил-1,4-нафтохинон. Витамин К содержится в зеленых травах, листьях, овощах. Предст. собой желтое масло, нераствор. в воде; перегоняется в высоком вакууме. Некоторые производные хинонов играют важную роль в промежуточных процессах биологического окисления.Антрахиноны – производные антрацена, содержащие хиноидное ядро. Антрахинон можно легко получить при окислении антрацена азотной кислотой или хромовой смесью. При этом в молекуле образуются две кето-группы и среднее кольцо приобретает строение хинона. Антрахинон представляет собой кристаллическое вещество желтого цвета, в отличие от обычных хинонов довольно стойкое к ряду химических воздействий, в частности к окислению. Антрагидрохинон является промежуточным продуктом при восстановлении антрахинона в антрацен. Антрагидрохинон в свободном виде представляет собой кристаллы коричневого цвета. Имея два фенольных гидроксила, антрагидрохинон растворяется в щелочах; образующееся вещество типа фенолята обладает ярко-красным цветом. Антрахинон способен бромироваться, нитроваться и сульфироваться.Ализарин представляет собой 1,2-диоксиантрахинон.Эмодины. В медицинской практике в качестве слабительных средств часто пользуются препаратами (настойками, отварами и т. д.) из алоэ, ревеня, крушины, листьев сенны и т. д. Действующими веществами этих растений, как оказалось, являются производные антрахинона, а именно – замещенные ди– и триокси-антрахинонов, содержащиеся в растениях частью в свободном виде, частью в виде эфиров и гликози-дов. Эти производные ди– и триоксиантрахинонов часто объединяют в группу эмодинов. Примером эмодинов может служить франгулоэмодин, являющийся 3-метил-1,6,8-триоксиантрахиноном. Франгулоэмо-дин содержится в крушине
50. Представление о фенольных антиоксидантах. Фенольные соединения в природе. Витамин Е. Флавоноиды. Антиоксидантами (АО) или антиокислителями принято называть соединения различной химической природы, способные тормозить или устранять свободно-радикальное окисление органических веществ молекулярным кислородом. В течение многих лет антиоксиданты широко используются для продления срока службы и улучшения эксплуатационных качеств полимерных и горюче-смазочных материалов, предотвращения окислительной порчи пищевых продуктов, жирорастворимых витаминов, кормов и косметических средств. Применение АО в этих областях дает огромный экономический эффект и позволяет сберегать значительные сырьевые ресурсы. Среди синтетических АО широкое распространение получили алкилированные фенолы, что обусловлено как сравнительной простотой их производства, так и комплексом ценных свойств: высокой эффективностью, малой токсичностью, универсальностью действия и возможностью изменять их свойства в широких пределах варьированием заместителей. Под фенольными антиоксидантами (ФАО) понимают любые соединения вида Ar(OH)n, в которых одна или несколько гидроксильных групп соединены с ароматическим ядром, причем молекула АО может содержать несколько фрагментов Ar(OH)n. Фенольные соединения способны оказывать влияние на многие физиологические процессы, проходящие в организме человека. Например, в составе растительных препаратов эти вещества (такие как кумарин, свойства которого еще мало изучены, рутин, флавоноиды) стимулируют деятельность коры надпочечников, благодаря чему надпочечники начинают активнее выделять гормоны группы глюкокортикоидов (вид гормонов выделяемых надпочечниками). Они обладают самыми разнообразными биологическими свойствами. Например, фенольные соединения листьев толокнянки, груши, брусники проявляют себя как антисептики.
Фенолокарбоновые кислоты- производные ароматических углеводородов, в молекуле которых атомы Н бензольного кольца замещаются на карбо(-СООН) или гидроксильные группы(-ОН) Фенольные соединения давно нашли свое применение в медицине, их применяют при лечении неврозов и коронарной недостаточности. Фенольные соединения обладают мочегонным, седативным, желчегонным и кровоостанавливающим действием. Флаваноиды, биофлавоноиды относятся к фенолам, это желто-красные пигменты растений. Их много и в пищевых, и в лекарственных растениях. Биофлавоноиды укрепляют капилляры, выступают как онкопротекторы, участвуют в выведении из организма солей тяжелых металлов, радионуклидов. В группу биофлавоноидов входят особые вещества, также проявляющие Р-витаминную активность и прочие свойства биофлавоноидов, которые называются антоцианы. По химической структуре антоцианы - это флавоновые гликозиды.Дубильные вещества- это полимерные фенольные соединения. В медицине они используются как вяжущие, противовоспалительные желудочно-кишечные средства.Самым известным дубильным веществом является танин.Его не стоит принимать внутрь: это вызовет расстройство пищеварения.Катехины (их также относят к биофлавоноидам) являются производными флавонолов и антоцианов. Кумарины - это ароматические вещества с запахом свежего сена. Кумарины - антикоагулянты, то есть препятствуют быстрому сворачиванию крови, как, например, производное кумарина дикумарол. Он является антивитамином К и применяется для профилактики и лечения тромбозов и тромбофлебитов.
51. Простые эфиры. Номенклатура, классификация. Расщепление кислотами. Простые эфиры - органические вещества, в которых молекулы содержат углеводородные радикалы, соединенные атомом кислорода. Записать это можно следующим образом: R–O–R’, где R и R' являются одинаковыми или различными радикалами. Простые эфиры рассматриваются в качестве производных спиртов. Эти соединения имеют составные названия. При этом используется название радикалов (по возрастанию молекулярной массы) и, собственно, слово "эфир" (диметиловый эфир СН3ОСН3, метилэтиловый эфир С2Н5ОСН3 и так далее)
Номенклатура простых эфиров Согласно тривиальной номенклатуре простые эфиры называют по радикалам, связанным с атомом кислорода, добавляя слово "эфир".

По номенклатуре ИЮПАК эфиры рассматривают как алкоксиалканы. Корень слова определяет самая длинная алкильная группа.

Простые эфиры относятся к числу малореакционноспособных веществ и стабильны по отношению ко многим реагентам, но они чувствительны по отношению к кислороду и легко образуют взрывчатые гидроперекиси, которые являются причиной взрыва при неосторожном обращении.
1. Кислотное расщепление простых эфиров
Простые эфиры расщепляются при нагревании до 120-150о с концентр. водной 48% HBr или HI. В столь же жестких условиях расщепляются простые эфиры фенолов.

Однако эфиры, содержащие третичную алкильную группу, расщепляются очень легко.

Кислотное расщепление простых эфиров следует рассматривать как реакцию нуклеофильного замещения у насыщенного атома углерода. В зависимости от природы алкильных групп, связанных с кислородом, реализуется либо SN1, либо SN2- механизмы. Если эфир содержит первичные или вторичные алкильные группы, реализуется SN2- механизм, в котором бромид- или иодид-ион атакует протонированную форму эфира по менее замещенному атому углерода. В этом случае расщепление отличается высокой региоселективностью и, как правило, образуется только один их двух возможных спиртов (вторичный) и первичный алкилгалогенид.

Хлорид- и фторид-ионы в воде сильно сольватированы за счет водородных связей и обладают недостаточной нуклеофильностью для кислотного расщепления простых эфиров по SN2- механизму.
Простые эфиры с третичной алкильной, бензильной или аллильной группами реагируют по SN1- механизму с образованием карбокатиона в качестве интермедиата. Эти реакции идут в мягких условиях, а в качестве кислотного агента можно использовать трифторуксусную кислоту.

В препаративном отношении гораздо более удобными реагентами для расщепления эфиров являются BCl3 или BBr3. В этих случаях расщепление проходит уже при -20оС. Это особенно необходимо при наличии других функциональных групп или тогда, когда возможна изомеризация углеродного скелета.

Образование гидропероксидов, их обнаружение и разложение.
Гидропероксиды — это первые молекулярные продукты окисления углеводородов. Звено цепи при их образовании имеет вид:
 взаимодействие пероксидного радикала с углеводородом — определяет строение образующегося гидропероксида и последующих продуктов окисления. При этом соблюдается обычный для радикальных реакций порядок изменения реакционной способности атомов водорода, определяемый относительной стабильностью промежуточного радикала
взаимодействие пероксидного радикала с углеводородом — определяет строение образующегося гидропероксида и последующих продуктов окисления. При этом соблюдается обычный для радикальных реакций порядок изменения реакционной способности атомов водорода, определяемый относительной стабильностью промежуточного радикала  . Вследствие этого преимущественным местом атаки молекулы при окислении алканов становится -положение боковой цепи по отношению к ароматическому ядру, а для олефинов — алкильное положение. Кроме того, для углеводородов всех классов справедлива известная последовательность в изменении способности к замещению атомов водорода, находящихся при разных углеродных атомах (третичный вторичный первичный).
. Вследствие этого преимущественным местом атаки молекулы при окислении алканов становится -положение боковой цепи по отношению к ароматическому ядру, а для олефинов — алкильное положение. Кроме того, для углеводородов всех классов справедлива известная последовательность в изменении способности к замещению атомов водорода, находящихся при разных углеродных атомах (третичный вторичный первичный).
Гидропероксиды относятся к числу довольно нестабильных соединений, превращающихся при окислении в другие продукты. Гидропероксиды при разложении под действием повышенной температуры или катализаторов окисления дают спирты и карбонильные соединения. Это разложение может иметь молекулярный механизм, однако в развившемся процессе окисления продукты образуются главным образом цепным путем. При получении спиртов звено цепи таково:


Кетоны образуются из вторичных гидропероксидов через стадию радикал-гидропероксидов:

Третичные гидропероксиды при цепном превращении дают кроме спирта с тем же числом углеродных атомов также спирт и кетон с меньшим числом атомов углерода за счет деструкции углерод-углеродной связи:


Особенности свойств ариламинов. Реакции электрофильного замещения в бензольном ядре ариламинов и их производных. Реакции диазотирования, соли арилдиазония. Реакции солей арилдиазония с выделением азота и без выделения азота.
Для арилами но в характерны реакции с участием атома азота и реакции с участием атомов углерода ароматического ядра. Основность. Ароматические амины обладают основным характером. Однако они слабее, чем амины жирного ряда и даже слабее аммиака. Снижение основности обусловлено сопряжением неподеленной пары электронов атома азота с л:-электронной системой ароматического ядра. Анилин не образует соли с Н2СОг На основность ариламинов существенное влияние оказывают заместители в бензольном кольце. Электронодонорные заместители увеличивают основность, а электроноакцепторные — уменьшают ее. При перехоле от первичных к третичным основность ароматических аминов снижается.
Электрофильное замещение в бензольном кольце.
В реакциях электрофильного замещения в бензольном кольце атом водорода замещается на электрофильный реагент при сохранении ароматического характера исходного соединения.
Соли арендиазония образуются при взаимодействии первичных ароматических аминов с азотистой кислотой. В промышленности соли арендиазония нашли широкое применение для получения разнообразных азокрасителей всех цветов и оттенков. По этой причине диазотирование относится к числу важнейших и наиболее изученных реакций в органической химии.
Диазотирование первичных ароматических аминов описывается следующим суммарным уравнением:
ArNH2+ NaNO2+ 2 HCl ArN+=N Cl - + NaCl + 2 H2O
ArN+=N Cl - + NaCl + 2 H2O
Реакции с выделением азота. При кипячении кислых растворов солей диазония происходит выделение азота и получаются фенолы. Превращение солей диазония без выделения азота. Реакции этой группы делают возможным переход от диазосоединений к азосоединениям (производным азобензола).Органические вещества этого класса лежат в основе одного из разделов промышленности, производящей синтетические красители из продуктов, добываемых из каменноугольного дегтя. Все азокрасители получаются при помощи так называемой реакции сочетания солей диазония.
59.Карбонильные соединения. Классификация, номенклатура и изомерия карбонильных соединений. Органические соединения, в молекуле которых имеется карбонильная группа >С=O, называются карбонильными соединениями, или оксосоединениями. Карбонильные соединения делятся на две большие группы — альдегиды и кетоны.
Альдегиды содержат в молекуле карбонильную группу, связанную с атомом водорода, т. е. альдегидную группу —СН=O. Кетоны содержат карбонильную группу, связанную с двумя углеводородными радикалами, т. е. кет