ГЕПАТОЦЕРЕБРАЛЬНАЯ ДЕГЕНЕРАЦИЯ.
ГЕПАТОЦЕРЕБРАЛЬНАЯ ДИСТРОФИЯ.
ЧАСТЬ 1.
МЕДЬ.
- могучий минерал, один из важнейших незаменимых микроэлементов. Она не только совершенно необходима, чтобы помогать сердцу правильно функционировать, но также контролирует уровни холестерина, сахара и мочевой кислоты.
Организм здорового человека очень четко регулирует ее количество: при избытке активно выводит, при недостатке экономит. Такая точность имеет причину: медь содержится в нашем теле в ничтожном количестве, мы не имеем стратегических запасов, так что нехватка сразу сказалась бы на течении многих процессов. А процессы, для которых требуется медь, – самые наиважнейшие. К примеру, вместе с железом она участвует в процессе образования эритроцитов – красных клеток крови.
медь – важный компонент костной ткани. Поэтому в периоды активного роста, во время беременности, а также при разного рода "испытаниях на прочность" она расходуется особенно активно.
Медь контролирует здоровье костей и сдерживает рост молочницы и других патогенных грибов.
Медь – важнейший компонент коллагена, основного нашего структурного белка. В частности, она участвует в создании сетки из эластиновых и коллагеновых волокон. От того, насколько хорошо сплетена эта сетка, зависит упругость и прочность как нашей кожи, так и сосудов.
Еще одна заметная функция меди – участие в образовании пигментов кожи и волос. Когда вы лежите на пляже, организм в авральном режиме производит пигмент меланин для защиты от повреждающего действия солнечных лучей. Это называют загаром. А поскольку поверхность тела весьма обширна, то на эту защиту уходит значительная часть того небольшого количества меди, которое имеется в организме здорового человека.
В сутки, любому человеку, нужно около 2,5 мг меди: меньше – дефицит, больше – яд, поэтому крайне важно поддерживать правильный баланс этого минерала. Как недостаток, так и избыток меди могут усиливать активность свободных радикалов, тем самым повышая риск болезней сердца и других хронических дегенеративных заболеваний.
Медь может существовать в двух состояниях (Cu+1 и/или Cu+2). В результате этого медь может вступать в реакцию с различными ферментами, а также участвовать в реакциях, в результате которых возникают активированные кислородом вещества.
Избыточная медь представляет собой высокотоксичный яд. Заболевания, связанные с метаболизмом меди, происходят в тех случаях, когда в организме контроль меди, отлагающейся в тканях, по какой-либо причине ослабевает. Существует две болезни людей, связанные с транспортом меди. Эти болезни возникают в результате нарушения транспорта металлической меди, осуществляемого аденозин трифосфотазами (ATP) Р-типа.
В нашем фильме речь пойдет о болезни Вильсона-Коновалова, которое является аутосомальным рецессивным заболеванием, в результате которого происходит отложение меди в мышечной ткани, печени и других органах.
ЧАСТЬ 2.
ИСТОРИЯ
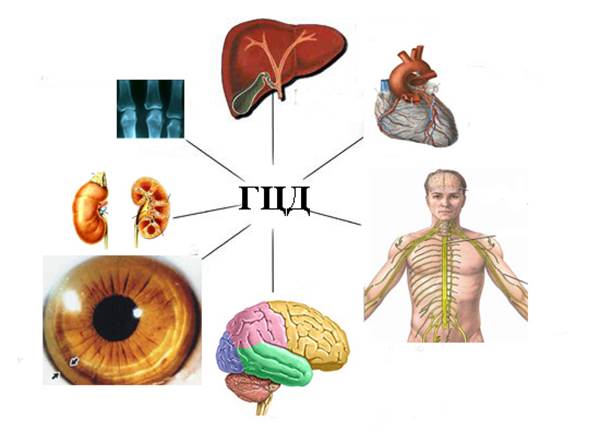
Гепато-церебральная дистрофия гепато-лентикулярная дегенерация, псевдосклероз Вестфаля, болезнь Вильсона-Коновалова – все это синонимы одного наследственного заболевания которое характеризуется сочетанием цирроза печени с дистрофическим процессом в головном мозге (преимущественно в чечевичных ядрах).В международном классификаторе болезней заболеванию присвоен код Е 83.0.

В 1912 году американский невролог Вильсон (S. Wilson), работавший в англии, описал болезнь, как наследственное фамильное заболевание, типичные для гепато-церебральной дистрофии изменения в головном мозге, установил постоянное наличие цирроза печени и дал описание клиники нового заболевания, названного им прогрессивной лентикулярной дегенерацией (лат. lenticularis чечевицеобразный), а так же заметил связь с появление “колец” (темного бурого пигмента в роговице глаз которые описали Кайзер (Kayser В)., в 1902; и Флейшер (Fleischer R.), в 1903году. В качестве основных симптомов заболевания были отмечены разнообразные непроизвольные движения в конечностях и туловище, мышечная ригидность, приводящая к скованности, дисфагия и дизартрия, аффектные вспышки, иногда психические расстройства, но признаки поражения пирамидных путей отсутствовали.
Ещё раньше К. Вестфалем в 1883году и А. Штрюмпеллем в 1898году было описано заболевание, которое по клиническому сходству с рассеянным склерозом получило название "псевдосклероз". Заболевание характеризовалось распространёнными, размашистыми, ритмичными непроизвольными движениями, повышением мышечного тонуса, амимией, дизартрией и выраженными психическими нарушениями вплоть до такого расстройства интеллекта, как слабоумие. В дальнейшем оказалось, что прогрессивная лентикулярная дегенерация и псевдосклероз являются разными формами одного и того же заболевания, которое Галль в 1921 назвал гепато-лентикулярной дегенерацией. Однако изменения в мозге при нем никогда не ограничиваются лентикулярными ядрами и нередко бывают даже сильнее выражены в других отделах мозга.

Николай Васильевич Коновалов Академик Академии Медицинских Наук СССР, руководил Научно-Исследовательским Институтом Неврологии с 1948 г. по 1966 г. Выдающийся русский ученый-энциклопедист, занимался изучением гепатолентикулярной дегенерации, впоследствии получившей название болезни Вестфаля-Вильсона-Коновалова.
в 1960 году предложил название "гепато-церебральная дистрофия" ГЦД. Коновалов выявил наличие морфологических повреждений всех структур головного мозга, а не только чечевицеобразных ядер.Он значительно расширил представления о патофизиологии, патогенезе и клинике этой болезни и выделил основные её формы.
5 основных Форм ГЦД по Коновалову:
• 1. Брюшная форма (доневрологическая) – манифестирует в возрасте от 5 до 17 лет и характеризуется различными вариантами поражения печени, нередко принимающими злокачественное «галопирующее» течение.
•
2. Аритмогиперкинетическая форма – возникает в раннем возрасте (от 7 до 15 лет) и проявляется полиморфными инвалидизирующими экстрапирамидными гиперкинезами (чаще торсионно-дистонического характера), интеллектуальным снижением и тяжелыми висцеральными расстройствами. Без лечения летальный исход наступает через 2–3 года.
• 3. Дрожательно-ригидная форма возникает в возрасте от 15 до 25 лет и отличается более доброкачественным течением. Она встречается чаще других и ближе всего соответствует форме, описанной Вильсоном. Характерно одновременное развитие ригидности и дрожания. Без лечения заболевание прогрессирует в течение 5–6 лет и заканчивается летально.
• 4. Дрожательная форма (соответствующая форме Вестфаля) отличается более поздним началом (в среднем в возрасте 20–30 лет) и наиболее доброкачественным (10–15 лет) течением. Известны случаи дебюта заболевания в 40 и даже в 50 лет. Основным клиническим симптомом является дрожание. Висцеральные проявления выражены менее значительно.
• 5. Экстрапирамидно-корковая форма представляет собой финал перечисленных выше форм, может развиться в течение времени или под действием внешних факторов (например, черепно-мозговой травмы) из любой основной неврологической формы ГЛД. Характеризуется присоединением к имеющимся типичным нарушениям остро развивающихся пирамидных парезов и эпилептических пароксизмов, чаще очагового характера. Быстро прогрессируют психические нарушения.
ЧАСТЬ 3.
ЭТИОЛОГИЯ
Гепатолентикулярная дегенерация известна с глубокой древности. Дошедшее до нас изображение египетского фараона Тутанхамона, по мнению крупнейшего специалиста J. Walshe, занимающегося всю жизнь проблемой гепатолентикулярной дегенерации, не исключает предположения, что он страдал этим заболеванием.

Это наследственное заболевание, характеризующееся поражением нервной системы и печени, передаётся при наличии, у обоих родителей генетической мутации в 13 хромосоме в ATP7B гене. Ген БВК, идентифицированный в 1993г, независимо сразу в 2х лабораториях США, представляет собой медь-транспортирую- щую АТФазу P типа с 6-ю металл связывающими районами. Ген имеет 60% гомологию по нуклеотидному составу с ранее идентифицированным геном АТФ-азы (АТР7А), мутантном при болезни Менкеса (Bull et al., 1993; Petruchin et al., 1993; Tanzi et al., 1993). По аналогии с геном болезни Менкеса, также обусловленной нарушением транспорта меди, ген БВК назван АТР7В. Два пациента с ГЦД оказались гомозиготными по 7-нукле- отидной делеции в кодирующей области гена ATP7B, что доказывало его идентичность гену ГЦД (Petruchin et al, 1993). При генетическом исследовании ГЛД, отмечается дефект в локусе 13g14.3 тринадцатой хромосомы.

Заболевание наследуется по аутосомно-рецессивному типу. При аутосомно-рецессивном типе наследования мутантный ген реализуется в признак в гомозиготном состоянии. Гетерозиготы клинически не отличаются от здоровых лиц. У фенотипически здоровых родителей, но имеющих рецессивный ген патологического признака, вероятность рождения больных детей составит 25 %, еще 25 % будут здоровы и фенотипически и генетически, а оставшаяся половина окажутся гетерозиготными носителями патологического признака, как и их родители. Вероятность заболевания мальчиков и девочек одинаковая.

В родословной при аутосомно-рецессивном наследовании заболевание может проявляться через одно или несколько поколений, при условии брака гетерозигот с гетерзиготами. Браки гетерозигот (здоровых) с гомозиготами (больными) встречаются в основном среди кровнородственных браков. Вероятность рождения больных детей при этом возрастает до 50 %. Браки, когда оба родителя гомозиготны достаточно редки. Все дети в этих семьях будут гомозиготами, а потому больными.
В 1960 году аутосомно-рецессивный тип наследственности был подтвержден. В 1985 году было установлено, что дефектный ген находится в 13 хромосоме. В 1993 году дефектный ген, ответственный за возникновение болезни Вильсона, был идентифицирован. Ген ATP7B, иногда называемый WND, кодирует транспортирующую металл ATPase (аденозин-трифосфотазу) ATP7B - ATPase Вильсона, в котором имеется 6 медьсвязывающих ветвей (motifs). Аминовый кончик ATPase Вильсона, включая все блоки связывания меди, был выделен в 1997 году, при этом была полностью описана медьсвязывающая функция данного домена. ATPase Вильсона была описана путем гомологического моделирования в 2002 году. Так как дефектный ген у больных, страдающих болезнью Вильсона, был обнаружен более 12 лет назад, на сегодняшний день удалось за это время описать более 350 различных вариаций мутаций, приводящих к развитию ГЛД. Распространенность любой из них указывает на время появления: чем чаще встречается мутация, тем она древней (возникла на ранних этапах формирования человечества). Для русских наиболее характерна мутация, распространенная среди европейских народов. В некоторых этнических группах, наряду с широко известными, можно обнаружить “молодые” мутации, свойственные только им. Например, у представителей татарской национальности выявлена мутация, которая нигде больше не встречается. Молекулярно-генетический анализ позволил предположить ее тюркские корни.
Таким образом, частота возникновения болезней, наследуемых по аутосомно-рецессивному типу, зависит от концентрации рецессивного гена в популяции и находится в прямой зависимости от степени распространения мутантного гена. География болезни Вильсона охватывает весь мир. Частота этого заболевания составляет около 10 случаев на миллион. Особенно повышается частота заболевания в изолятах и среди населения с высоким процентом кровнородственных браков. Например, у жителей Сардинии и китайцев. Однако, болезнь Вильсона достаточно редко встречается у жителей Африки. Заболевают лица любой этнической и географической принадлежности. Гетерозиготное носительство дефектного гена достигает частоты 1к100.
В каждой клетке человека имеются 23 пары хромосом.
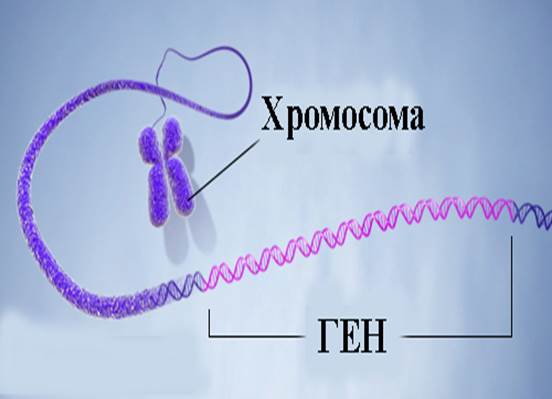
22 пары одинаковы у женщин и мужчин. 23-я пара у женщин содержит две одинаковые хромосомы (их называют Х-хромосомами), а у мужчин - две разные хромосомы (Х-хромосому и Y-хромосому). Эти хромосомы, по которым отличаются между собой мужчины и женщины, называются половыми. В половые клетки попадает только хромосома из каждой пары. Поэтому мужские половые клетки у человека несут в себе или Х-хромосому, или Y-хромосому. От того, с какой из этих клеток сольётся при оплодотворении женская половая клетка (всегда несущая Х-хромосому), будет зависеть пол зародыша. Женщина получает две Х-хромосомы: одну от отца и одну от матери. Мужчина получает одну Х-хромосому от матери и одну Y-хромосому от отца.

*Локус в биологии означает фиксированное положение (локализацию) на хромосоме, например положение гена. Вариант последовательности ДНК в данном локусе называется аллелью. Упорядоченный перечень локусов для какого-либо генома называется генетической картой. Генное картирование — это определение локусa для специфического биологического признака.
Диплоидные или полиплоидые клетки, которые несут одинаковые аллели на каком-либо локусе называются гомозиготными по этому локусу, а те, которые несут различные аллели — гетерозиготными.
Заболевание поражает около 25% братьев-сестер в каждой конкретной семье при клинически здоровых родителях (аутосомно-рецессивный тип наследования). Каждый из родителей является носителем одной копии патологического гена (их называют гетерозиготными носителями мутации). При таком типе наследования заболевают только те лица, которые унаследовали от родителей два патологических гена - от матери и от отца (такие лица называются гомозиготными носителями мутации).
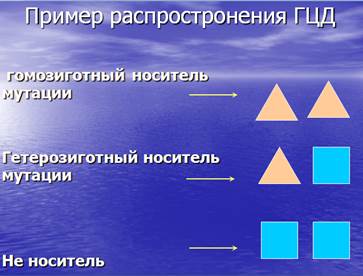
При наличии в семье одного клинически больного родителя гомозиготного
носителя гена АТР7В, все дети будут являться гетерозиготными носителями.

При наличии в семье двух клинически здоровых родителей гетерозиготных
носителей гена АТР7В, вероятность рождения больного гомозиготного ребёнка составляет 25-50%.

При наличии в семье двух родителей один из которых гетерозиготный носитель гена АТР7В, а другой гомозиготный, вероятность рождения больного гомозиготного
ребёнка составляет 75%.

К сожалению, до настоящего времени не установлен точный механизм и генетические причины данного заболевания. Пока не удалось установить четкой связи между нарушениями в гене человека и печеночным токсикозом, вызываемым медью, отлагающейся в тканях. Но известны Типичные молекулярно-генетические изменения при ГЛД.
• Мутантный ген, ведущий к нарушению синтеза функции первичного белка: медной АТФ-азы Р типа, локализующейся на 13-ой хромосоме (13q14.3).
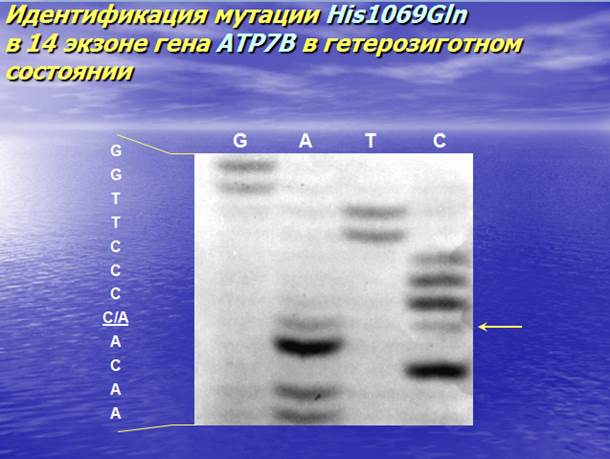
• Мажорной мутацией, по данным НИИ неврологии РАМН, польских и сербских исследователей (славянская популяция) (60-70% исследованных больных) имеют мутацию в 14-ом экзоне – His1069Gln (замена гистидина на глутамин).
• В различных странах выявлены мутации в 15-ом экзоне (Россия, 3%), 8-ом экзоне (Россия, 1%).
• ДНК-диагностика в доклинической стадии наряду с низкой концентрацией церулоплазмина в сыворотке крови дает возможность точной диагностики ГЛД на ранних стадиях болезни.
ЦЕРУЛОПЛАЗМИН (Сеruloplasminum). В 1944 году церулоплазмин (ЦП) был впервые описан шведскими учеными Холмбергом и Лауреллом [Holmberg, 1944], и подробно охарактеризован спустя несколько лет. Тогда же он получил свое название, означающее “небесно-голубой белок плазмы” [Holmberg & Laurell, 1948; Holmberg & Laurell, 1951]. После того, как была подробно изучена способность ЦП окислять Fe2+ до Fe3+ и доказано физиологическое значение этой реакции [Osaki, 1966], белок получил также систематическое название согласно Международной Классификации Ферментов - “ферро-О2-оксидоредуктаза, КФ 1.16.3.1”. Часто используется более короткое название - ферроксидаза. Гликопротеид a-глобулиновой фракции, является белком плазмы крови, выполняющим в организме ряд важных биологических функций; повышает стабильность клеточных мембран, участвует в иммунологических реакциях, ионном обмене, оказывает антиоксидантное действие, тормозит перекисное окисление липидов, стимулирует гемопоэз (Процесс размножения, развития и созревания клеток крови путем последовательных дифференциаций(повторений) из исходной стволовой клетки). Точная локализация этого протеина в гепатоците неизвестна, однако вероятнее всего он участвует в переносе меди из лизосом гепатоцитов в желчь.
При редкой атипичной форме, предположительно Германского происхождения, у гетерозигот содержание церулоплазмина снижено, по крайней мере, в два раза. При двух других, типичных формах - славянской и ювенильной, содержание церулоплазмина у гетерозигот находится в пределах нормы. Славянский тип ГЦД характеризуется сравнительно поздним началом и преимущественно неврологической симптоматикой. Ювенильная форма чаще встречается в Западной Европе и ведущими в этиологии заболевания являются печеночные нарушения. Среди евреев-ашкенази встречается БВК с поздним началом и почти нормальным содержанием церулоплазмина в сыворотке крови больных.
До 4-6 лет в организме присутствует так называемый эмбриональный церулоплазмин поэтому постепенное накопление меди происходит после 4х летного возраста. Организм человека включает различные защитные механизмы направленные на то чтобы максимально компенсировать поражение. Этот фактор и различные вариации мутаций определяют время и форму манифестации заболевания.
ЧАСТЬ 4.
ТРАНСПОРТ МЕДИ
Обычно среднесуточное потребление пищи включает в себя значительное количество меди. При этом поступление меди в организм варьируется в пределах от 1 до 10 мг в сутки.

Как правило, от 2 до 5 мг в сутки поступает с мясными продуктами, бобовыми, ракообразными, а также с шоколадом. Рекомендуемее поступление меди в организм при ГЦД составляет примерно 0,9 мг в сутки. Большая часть меди (до 85%), поступающей с пищей, выводится, и только 15% удерживается в тканях тела. Главным образом, выделение меди, поступающей с пищей, происходит по гепатобиллиарной схеме. При таком механизме отсутствует циркуляция меди между кишечником и печенью (enterohepatic). Как правило, с мочой выводится менее 5% меди, если только способность обратного всасывания через канальцы почек не превышена, как это происходит при ГЦД. Таким образом, регулирование выделения меди в желчь очень важно для поддержания нормального гомеостаза меди в теле человека. Медь, поступающая с пищей, а также медь, содержащаяся в желчи, слюне, желудочном соке и секрете поджелудочной железы абсорбируется в тонкой кишке, 12-типерстной кишке и проксимальной части тощей кишки. Абсорбция меди происходит, вероятно, посредством CTRI, проявляющегося на энтероцитах, несмотря на наличие двухвалентного транспортного катиона (DMTI), который отвечает, главным образом, за перенос железа, и в силу этого, вероятно, играет незначительную роль в транспорте меди. Абсорбированная медь связывается обратимо с альбумином сыворотки и с различными аминокислотами, среди которых наиболее важной является гистидин.
Содержащие медь альбумин и гистидин перераспределяют медь между различными тканями организма (главным образом, направляя ее в ткани печени). Медь, свободно прикрепленная к аминокислотам, фильтруется в почках, и реабсорбируется в канальцах. Для функционирования основных ферментов (таких, как лизилоксидаза), участвующих в формировании соединительной ткани и эластических поперечных связок, необходимы лишь незначительные количества меди (следы). Сюда же следует отнести супероксид Cu/Zn дисмутаза (SODI), одной из функций которого является очистка цитоплазмы от свободных радикалов, цитохром С-оксидазу, ответственную за электронный перенос митохондрий, тироксиназу, необходимую для производства пегмента, допамин b-моноксигеназу, ответственную за процесс нейротрансмиссии, а также пепсидил a-аминирующую монооксидазу, ответственную за процесс нейропередачи сигналов. Молекулярная медь не присутствует в свободном состоянии внутри клеток. В гепатоцитах и других клетках медь находится всегда в виде соединений с низкомолекулярными протеинами, так называемыми металлошаперонами (спутниками металлов), каждый из которых поставляет медь к различным молекулам, находящимся внутри клетки, что необходимо для осуществления синтеза новых протеинов (белков). В гепатоцитах медь объединяется с фероксидазой церулоплазмина c молекулярным весом 132 kDа, являющейся гликопротеином с шестью молекулами меди. Церулоплазмин окисляет железо от феритина до трансферина. Церулоплазмин, в котором имеется дефицит меди (апоцерулоплазмин), не участвует в процессе, в котором задействована феррооксидаза. Продолжительность существования церулоплазмина в плазме довольно невелика, и составляет около 24 часов. При попадании меди в апоцерулоплазмин образуется ферментоактивный протеин-церулоплазмин (известный под названием - голоцерулоплазмин).

ATPase Вильсона, вероятно, является основным продуктом, необходимым для синтеза церулоплазмина. Примерно 95% меди в плазме находится в церулоплазмине. В связи с этим, дальнейший обмен меди невозможен. Как энтероциты, так и гепатоциты содержат металлотионеины – вещества, относящиеся к классу низкомолекулярных протеинов, обогащенных цистеином. Эти вещества все время производятся в организме и могут переводить медь в нетоксические формы. Медь также может образовывать комплексы с интроцеллюлярными глютатионами.
ЧАСТЬ 5.
Патогенез
В норме практически вся тканевая медь находится в простетических группах медьсодержащих ферментов, таких как цитохромоксидаза, тирозиназа, супероксиддисмутаза и церулоплазмин. Свободной (не связанной с белком) меди в норме мало или совсем нет. При болезни Вильсона в организме количество меди больше, чем способно связаться специфическими медьсодержащими белками. Она столь же токсична, как и находящиеся в избытке железо, цинк, ртуть или свинец. Токсичность этих катионов в большей степени объясняется, их патологическим связыванием белками, в норме не содержащими металлов.
В основе патогенеза гепатолентикулярной дегенерации лежат следующие механизмы:
n Мутация медной АТФ-азы Р –типа с последующим нарушение выведения из печени фракции меди с желчью, так называемой регуляторной меди, поддерживающей в физиологических условиях отрицательный баланс меди в организме;
n Вторично обусловленное снижение скорости включения меди в церулоплазмин, обладающий транспортной функцией
n Постепенное токсическое депонирование и поражение органов-мишеней (печень-почки-мозг). 

n Присоединение энцефалопатии смешенного типа - гепатогенная энцефалопатия + специфическое поражение подкорковых структур имеющих высокую афинность к ионам меди.
Участки отложения меди видны на снимках компьютерной и магнито-резонансной томографии

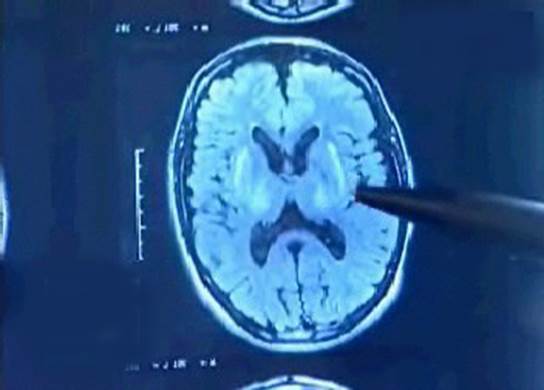




n В мозге избыток меди определяют постоянно. Некрозу нейронов с образованием полостей может предшествовать появление клеток Опальского или Альцгеймера II типа, однако они неспецифичны для болезни Вильсона.
n Развитие аутоиммунных реакций с появлением повышенных титров аутоантител (а-АТ) к нейроспецифическим белкам, с последующей аутоиммунной агрессией и прорывом ГЭБ.
n В патогенезе этой болезни ведущую роль играет нарушение экскреции меди с желчью вследствие дефекта гена, который кодирует продукцию в гепатоцитах специфической АТФ-азы, определяющей транспорт меди в аппарат Гольджи и ее последующее выделение с лизосомами в желчь. При этом в крови происходит снижение уровня церулоплазмина (медьвыводящего белка).
Все это, предположительно, ведет к нарушению синтеза церулоплазмина и снижению его концентрации в сыворотке крови.
Цитотоксический эффект свободной меди на уровне поражения органов определяет клиническую симптоматику со стороны ---центральной нервной системы (неврологические полиморфные расстройства)
внутренних органов--- в первую очередь в печени (Вильсоновский гепатит переходящий в цирроз печени)
Примерно в половине случаев болезнь Вильсона проявляется печеночной патологией. Токсические эффекты меди в печени обнаруживают себя острым гепатитом, циррозом или бессимптомной гепатоспленомегалией. Острый гепатит напоминает вирусный, его можно спутать с инфекционным мононуклеозом, и он может развиваться в трех разных направлениях. Первое — это скоротечная, иногда летальная форма болезни, проявляющаяся желтухой, недомоганием и подчас асцитом, гипоальбуминемией и повышением уровня печеночных ферментов в плазме. В острой фазе в плазму может поступать достаточное для индукции гемолитической анемии количество меди. Диагноз иногда не удается поставить до аутопсии или до того момента, когда заболевание младших сиблингов заставит произвести анализ сохранившихся тканей на медь. Второе направление — это постепенное повреждение паренхимы печени, обусловливающее клинические и гистологические изменения, неотличимые от таковых при хроническом активном гепатите. Третье — внешнее выздоровление больного после гепатита, несмотря на развитие в дальнейшем цирроза печени. До появления признаков или симптомов болезни может пройти несколько лет и даже десятилетий. В этих случаях без тщательного опроса больного или при отсутствии клинически выраженного цирроза можно и не обратить внимания на имевший место в прошлом приступ гепатита.
После того как печень насыщается медью, металл накапливается в центральной нервной системе и, в частности, в подкорковых ядрах головного мозга, что ведет к нейропсихическим нарушениям (тремор, дизартрия, атаксия, снижение интеллекта, изменения личности, судороги). Кроме того, депонирование меди происходит в других органах и тканях (почках, коже, сердце, эндокринных железах), Токсическое поражение Центральной Нервной Системы начинается с накопления меди в Мозгу.
Происходит также Изменения костно-суставной системы. Артропатия, которая наблюдается у 25–50% пациентов, чаще в возрасте старше 20 лет.

Вовлекаются крупные суставы – бедренный и коленный, суставы запястья и позвоночника. При рентгенологическом исследовании определяются дегенеративные изменения: остеофиты, склероз, подхрящевые псевдокисты и фрагментация кости.
Сопутствует также и анемия Фанкони (болезнь, характеризующаяся гипоплазией костного мозга, панцитопенией, аномалиями развития кожи, костной системы, внутренних органов), синдром Фанкони (болезнь, характеризующаяся гипоплазией костного мозга, панцитопенией, аномалиями развития кожи, костной системы, внутренних органов)
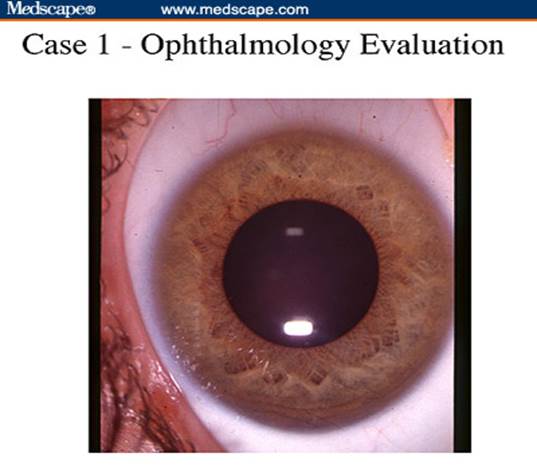
Скопление меди в десметовой мембране задней стенке глаз, ведет к формированию кольца Кайзера- Флейшера.

Эти зеленого или золотистого цвета отложения меди в десцеметовой оболочке роговицы не мешают зрению, но указывают на высвобождение меди из печени и повреждение мозга.

Отложение меди в роговице глаз, лучше выявляется при осмотре в щелевой лампе.

Иногда появлению колец сопутствуют подсолнечниковые катаракты.
Часто видны не вооруженным глазом.
ККФ у кариглозого



ККФ у голубоглазого



Кольцо Кайзера-Флейшера
у альбиноса.

ККФ у африканца

ККФ у черноглазого

ККФ у зеленоглазого

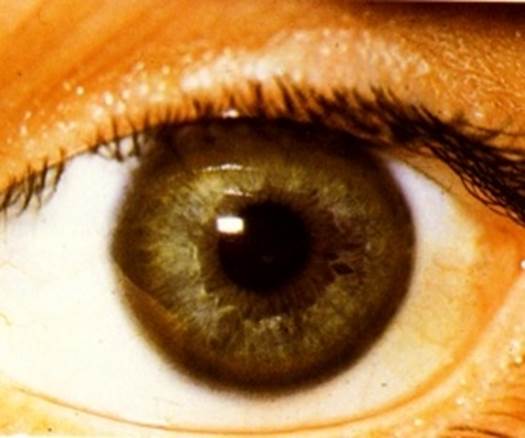
ККФ у сероглазого

Печень.

Печень - самый крупный из внутренних органов. Она контролирует многие обменные процессы, играющие важную роль в поддержании постоянного состава крови, и многие ее функции связаны с переработкой всасываемых компонентов пищи.
У человека печень имеет большие размеры и составляет 3 - 5% общей массы тела. Расположена она непосредственно под диафрагмой, к которой прикреплена серповидной связкой. Она состоит из нескольки долей, причем форма может изменяться в зависимости от количества находящейся в ней крови. Снаружи печень окружена оболочкой, состоящей из двух слоев: наружный слой образует гладкая влажная брюшина печени, а внутренний - фиброзная глиссонова капсула печени, которая окружает все структуры, входящие в печень и выходящие из нее. Волокна глиссоновой капсулы, расположенные внутри печени, поддерживают ее форму.
Клетки печени называются гепатоцитами. Они содержат крупное ядро, аппарат Гольджи, многочисленные клетки: митохондрии и лизосомы, а также множество гликогеновых гранул и липидных капелек. Они плотно примыкают друг к другу и на поверхности, обращенной к кровеносным капиллярам, имеют микроворсинки, через которые и происходит обмен веществами между гепатоцитами и кровью Кроме гепатоцитов в печени имеются нервные элементы и клетки, связанные с кровеносными и лимфатическими сосудами.
В целом внутреннее строение печени довольно сложно и до конца не изучено. Оно основано на определенном взаимном расположении гепатоцитов и двух систем кровеносных сосудов, переплетающихся с желчными канальцами печени (желчными капиллярами печени). В них имеются венулы, от которых отходят более мелкие кровеносные сосуды, называемые синусоидами, которые образуют густую сеть капиллярных сосудов и отделены друг от друга пластинками гепатоцитов толщиной в одну клетку. В синусоидах происходит обмен веществами между кровью и гепатоцитами. Этот обмен облегчается благодаря наличию в эндотелии синусоидов пор диаметром до 10 нм и микроворсинок на поверхности гепатоцитов, обращенной к синусоидам. Желчь, образующаяся в гепатоцитах, поступает в мельчайшие желчные капилляры, которые, перемежаясь с синусоидами, проходят между соседними слоями гепатоцитов. В желчные капилляры выступают микроворсинки гепатоцитов, через которые желчь выводится в капилляры путем активного транспорта. Желчные капилляры образуют разветвленную сеть и сливаются в мелкие желчные протоки, которые соединяются вместе в портальном тракте и образуют более крупные протоки, сливающиеся в общий желчный печеночный проток. Печень страдает при многих врожденных нарушениях обмена веществ, в том числе при болезнях накопления.
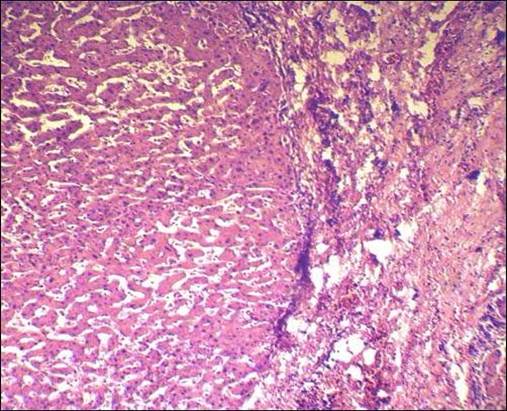
Цирроз печени у больного ГЦД