Наследственность и изменчивость - фундаментальные свойства живого, их диалектическое единство. Общее понятие о генетическом материале и его свойствах: хранение, репарация генетической информации, передача ее от поколения к поколению.
Наследственность – свойство живых организмов, обеспечивающее материальную преемственность онтогенеза в определенных условиях внешней среды. Гены детерминируют последовательность полипептидной цепи.
Наследование – передача информации от одного поколения к другому. Благодаря наследственности стало возможно существование популяций, видов и других групп.
1953 год – расшифрована структура молекулы ДНК.
Наследственность, присущее всем организмам свойство повторять в ряду поколений одинаковые признаки и особенности развития; обусловлено передачей в процессе размножения от одного поколения к другому материальных структур клетки, содержащих программы развития из них новых особей. Тем самым Н. обеспечивает преемственность морфологической, физиологической и биохимической организации живых существ, характера их индивидуального развития, или онтогенеза. Как общебиологическое явление Н. — важнейшее условие существования дифференцированных форм жизни, невозможных без относительного постоянства признаков организмов, хотя оно нарушается изменчивостью — возникновением различий между организмами. Затрагивая самые разнообразные признаки на всех этапах онтогенеза организмов, Н. проявляется в закономерностях наследования признаков, т. е. передачи их от родителей потомкам.
Изменчивость - это способность организма приобретать новые признаки в процессе онтогенеза. Изменчивость – свойство живых организмов существовать в разных формах
Формы изменчивости по характеру изменения признаков и механизму:
--фенотипическая:
- случайная
- модификационная
--генотипическая:
- соматическая
- генеративная (мутационная, комбинативная)
а) генная
б) хромосомная
в) геномная
Групповая и индивидуальная изменчивость – классификация по эволюционному значению. Изменчивость, реализованная группой организмов, называется групповой, у одного организма или группы его клеток – индивидуальная.
Значение генетики для медицины. Человек как специфический объект генетического анализа. Методы изучения наследственности человека.
Человек как: обьект генетического анализа
Трудности:
1. сложный кариотип
2. гетерозиготн6сть по многим генам
3. разнообразие среды
4. позднее половое созревание
5 малое число потомков
6 невозможность постановки экспериментов
Преимущества:
1. высокая численность доступна для изучения популяциии
2 значительное число и разнообразие известных мутаций и хромосомных аномалий
3 доскональные знания физиологии и биохимии человека
Методы исследования генетики: Генеалогический, близнецовый, цитогенетический, биохимический, популяционный.
Взаимодействие аллельных генов (доминирование, неполное доминирование, кодоминирование).
Полное доминирование — взаимодействие двух аллелей одного гена, когда доминантный аллель полностью исключает проявление действия второго аллеля. В фенотипе присутствует только признак, задаваемый доминантной аллелью.
Неполное доминирование — доминантный аллель в гетерозиготном состоянии не полностью подавляет действие рецессивного аллеля. Гетерозиготы имеют промежуточный характер признака.
Сверхдоминирование — более сильное проявление признака у гетерозиготной особи, чем у любой гомозиготной.
Кодоминирование — проявление у гибридов нового признака, обусловленного взаимодействием двух разных аллелей одного гена. Фенотип гетерозигот не является чем-то промежуточным между фенотипами разных гомозигот.
Взаимосвязь между геном и признаком. Пример. Гипотеза «один ген - один фермент», ее современная трактовка.
Открытия экзон-интронной организации эукариотических генов и возможности альтернативного сплайсинга показали, что одна и та же нуклеотидная последовательность первичного транскрипта может обеспечить синтез нескольких полипептидных цепей с разными функциями или их модифицированных аналогов. Например, в митохондриях дрожжей имеется ген box (или cob), кодирующий дыхательный фермент цитохром b. Он может существовать в двух формах (рис. 3.42). «Длинный» ген, состоящий из 6400 п. н., имеет 6 экзонов общей протяженностью 1155 п. н. и 5 интронов. Короткая форма гена состоит из 3300 п. н. и имеет 2 интрона. Она фактически представляет собой лишенный первых трех интронов «длинный» ген. Обе формы гена одинаково хорошо экспрессируются.
После удаления первого интрона «длинного» гена box на основе объединенной нуклеотидной последовательности двух первых экзонов и части нуклеотидов второго интрона образуется матрица для самостоятельного белка — РНК-матуразы (рис. 3.43). Функцией РНК-матуразы является обеспечение следующего этапа сплайсинга — удаление второго интрона из первичного транскрипта и в конечном счете образование матрицы для цитохрома b.
Другим примером может служить изменение схемы сплайсинга первичного транскрипта, кодирующего структуру молекул антител в лимфоцитах. Мембранная форма антител имеет на С-конце длинный «хвост» аминокислот, который обеспечивает фиксацию белка на мембране. У секретируемой формы антител такого хвоста нет, что объясняется удалением в ходе сплайсинга из первичного транскрипта кодирующих этот участок нуклеотидов.
У вирусов и бактерий описана ситуация, когда один ген может одновременно являться частью другого гена или некоторая нуклеотидная последовательность ДНК может быть составной частью двух разных перекрывающихся генов. Например, на физической карте генома фага ФХ174 (рис. 3.44) видно, что последовательность гена В располагается внутри гена А, а ген Е является частью последовательности гена D. Этой особенностью организации генома фага удалось объяснить существующее несоответствие между относительно небольшим его размером (он состоит из 5386 нуклеотидов) и числом аминокислотных остатков во всех синтезируемых белках, которое превышает теоретически допустимое при данной емкости генома. Возможность сборки разных пептидных цепей на мРНК, синтезированной с перекрывающихся генов (А и В или Е и D), обеспечивается наличием внутри этой мРНК участков связывания с рибосомами. Это позволяет начать трансляцию другого пептида с новой точки отсчета.
Нуклеотидная последовательность гена В является одновременно частью гена А, а ген Е составляет часть гена D
В геноме фага λ были также обнаружены перекрывающиеся гены, транслируемые как со сдвигом рамки, так и в той же рамке считывания. Предполагается также возможность транскрибирования двух разных мРНК с обеих комплементарных цепей одного участка ДНК. Это требует наличия промоторных областей,.определяющих движение РНК-полимеразы в разных направлениях вдоль молекулы ДНК.
Описанные ситуации, свидетельствующие о допустимости считывания разной информации с одной и той же последовательности ДНК, позволяют предположить, что перекрывающиеся гены представляют собой довольно распространенный элемент организации генома вирусов и, возможно, прокариот. У эукариот прерывистость генов также обеспечивает возможность синтеза разнообразных пептидов на основе одной и той же последовательности ДНК.
Имея в виду все сказанное, необходимо внести поправку в определение гена. Очевидно, нельзя больше говорить о гене как о непрерывной последовательности ДНК, однозначно кодирующей определенный белок. По-видимому, в настоящее время наиболее приемлемой все же следует считать формулу «Один ген — один поли-пептид», хотя некоторые авторы предлагают ее переиначить: «Один полипептид — один ген». Во всяком случае, под термином ген надо понимать функциональную единицу наследственного материала, по химической природе являющуюся полинуклеотидом и определяющую возможность синтеза полипептидной цепи, тРНК или рРНК.
Один ген один фермент.
В 1940 г Дж. Бидл и Эдвард Татум использовали новый подход для изучения того, как гены обеспечивают метаболизм у более удобного объекта исследований – у микроскопического грибка Neurospora crassa.. Ими были получены мутации, у которых; отсутствовала активность того-или иного фермента метаболизма. А это приводило к тому, что мутантный гриб бьл не способен сам синтезировать определенный метаболит (например, аминокислоту лейцин) и мог жить только тогда, когда лейцин был добавлен в питательную среду. Сформулированная Дж. Бидлом и Э. Татумом теория "один ген - один фермент" - быстро получила широкое признание у генетиков, а сами они были награждены Нобелевской Премией.
Методы. селекции так называемых "биохимических мутаций", приводящих к нарушениям действия ферментов, обеспечивающих разные пути метаболизма, оказались очень плодотворными не только для науки, но и для практики. Сначала они привели к возникновению генетики и селекции промышленных микроорганизмов, а потом и к микробиологической промышленности, которая использует штаммы микроорганизмов, сверх продуцирующие такие стратегически важные вещества, как антибиотики, витамины, аминокислоты и др.. В основе принципов селекции и генной инженерии штаммов сверхпродуцентов лежит представление, что "один ген кодирует один фермент". И хотя это представление отлично практике приносит многомиллионные прибыли и спасает миллионы жизней (антибиотики) - оно не является окончательным. Один ген - это не только один фермент.
Хромосомные мутации, их классификация. Причины и механизмы возникновения хромосомных мутаций. Роль хромосомных мутаций в развитии патологических состояний и эволюционном процессе. Хромосомные болезни человека. Примеры.
Нарушения структуры хромосом
Транслокации — обменные перестройки между негомологичными хромосомами.
Делеции — потери участка хромосомы. Например, синдром «кошачьего крика» связан с делецией короткого плеча 5-ой хромосомы. Признаком его служит необычный плач детей, напоминающий мяуканье или крик кошки. Это связано с патологией гортани или голосовых связок. Наиболее типичным, помимо «кошачьего крика», является умственное и физическое недоразвитие, микроцефалия (аномально уменьшенная голова).
Инверсии — повороты участка хромосомы на 180 градусов.
Дупликации — удвоения участка хромосомы.
Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах.
Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы.
Хромосомные мутации (хромосомные абберации) – структурные перестройки, затрагивающие одну или несколько хромосом. При всем многообразии структурных перестроек все они связаны с потерей либо с добавлением участка хромосомы. Частичные моносомии и трисомии (смотри 8 лекцию). На долю хромосомных мутаций приходится 7% хромосомных болезней. Клинически они сопровождаются множественными пороками развития и аномалиями.
Хромосомные болезни, наследственные заболевания, обусловленные изменением числа или структуры хромосом.
Эта группа заболеваний обусловлена изменением структуры отдельных хромосом или их количества в кариотипе. Как правило, при таких мутациях наблюдается дисбаланс наследственного материала, который и ведет к нарушению развития организма. У человека описаны геномные мутации по типу полиплоидии, которые редко наблюдаются у живорожденных, а в основном обнаруживаются у абортированных эмбрионов и плодов и у мертворожденных. Основную часть хромосомных болезней составляют анэуплоидии, причем моносомии по аутосомам у живорожденных встречаются крайне редко. Большинство из них касаются 21-й и 22-й хромосом и чаще обнаруживаются у мозаиков, имеющих одновременно клетки с нормальным и мутантным кариотипом. Достаточно редко обнаруживается моносомия и по Х-хромосоме (синдром Шерешевского — Тернера).
В отличие от моносомии трисомии описаны по большому числу аутосом: 8, 9, 13, 14, 18, 21, 22-й и Х-хромосоме, которая может присутствовать в кариотипе в 4—5 экземплярах, что вполне совместимо с жизнью.
Структурные перестройки хромосом также, как правило, сопровождаются дисбалансом генетического материала (делеции, дупликации). Степень снижения жизнеспособности при хромосомных аберрациях зависит от количества недостающего или избыточного наследственного материала и от вида измененной хромосомы.
К настоящему времени описано около 100 клинико-цитогенетических синдромов, в основе которых лежат различные хромосомные аномалии.
Хромосомные изменения, приводящие к порокам развития, чаще всего привносятся в зиготу с гаметой одного из родителей при оплодотворении. При этом все клетки нового организма будут содержать аномальный хромосомный набор и для диагностики такого заболевания достаточно проанализировать кариотип клеток какой-нибудь ткани.
Если хромосомные нарушения возникают в одном из бластомеров во время первых делений зиготы, образующейся из нормальных гамет, то развивается мозаичный организм, большая или меньшая часть клеток которого несет нормальный хромосомный набор. Диагностика мозаичных форм хромосомных болезней отличается большей трудоемкостью и требует изучения кариотипа большого числа клеток из разных тканей.
Для определения вероятности появления хромосомной болезни в потомстве в семьях, уже имеющих больных детей, важно установить, является ли это хромосомное нарушение заново возникшим или оно унаследовано от предыдущего поколения. Чаще родители человека с хромосомным заболеванием имеют нормальный кариотип, а появление больного потомства является результатом мутации, возникшей в одной из гамет. В этом случае возможность повторного хромосомного нарушения у детей в данной семье маловероятна и не превосходит таковой в целом для популяции. Вместе с тем описано немало семей, в которых наблюдается предрасположение, например, к нерасхождению хромосом.
В случае наследуемых хромосомных болезней в соматических клетках родителей обнаруживаются хромосомные или геномные мутации, которые могут передаваться их зрелым половым клеткам в ходе гаметогенеза. Передают потомству хромосомные нарушения обычно фенотипически нормальные родители, являющиеся носителями сбалансированных хромосомных перестроек — реципрокных транслокаций, робертсоновских транслокаций или перицентрических инверсий. У носителей такого рода хромосомных перестроек с определенной вероятностью образуются нормальные гаметы, а также гаметы, несущие сбалансированную перестройку, и половые клетки с нарушенным балансом генов в геноме (рис. 6.22).
Возможность наследования хромосомных аномалий делает необходимым анализ кариотипа родителей, уже имеющих больных детей, и пренатальную диагностику развивающегося внутриутробно плода для исключения вероятности повторного рождения ребенка с хромосомной болезнью.
Фенотипическое проявление различных хромосомных и геномных мутаций характеризуется ранним и множественным поражением различных систем органов. Типичными являются задержка общего физического и умственного развития, отклонения в строении скелета, в частности мозгового и лицевого черепа, пороки развития сердечно-сосудистой, мочеполовой, нервной систем, нарушения в биохимическом, гормональном и иммунологическом статусе организма. Хромосомные болезни, как правило, характеризуются сочетанием многих врожденных пороков. Для них также характерны многообразие и вариабельность фенотипических проявлений. Наиболее специфические проявления хромосомных заболеваний связаны с дисбалансом по относительно небольшому фрагменту хромосомы. Так, фенотипическое проявление синдрома Дауна наблюдается в случае трисомии всего лишь по небольшому сегменту длинного плеча 21-й хромосомы. Картина синдрома «кошачьего крика» развивается при утрате участка короткого плеча 5-й хромосомы. Дисбаланс по значительному объему хромосомного материала делает фенотипическую картину менее специфической.
Для медицинской практики в 1971 году был проведен симпозиум по медицинской генетике в Париже. Была принята международная Парижская классификация для обозначения кариотипа человека. 46,хх; 46,ху – кариотип нормального человека.
Во время мейоза возможно появление аномальных половых клеток.
47,хху – синдром Клайнфельтера.
Мужчина, частота встречаемости 1 из 1000 новорожденных мальчиков.
Высокий рост, более длинные ноги, евнуховидное телосложение, недоразвитие половых органов, гинекомастия, у половины умственная отсталость (трудности в обучении чтению и письму), могут заканчивать нормальные школы, хотя им может быть очень трудно. Вспыльчивы, импульсивны, легко попадают од влияние более сильных личностей, преступления и проступки. Жизнеспособность снижена. Среди «туповатых» преступников приблизительно 2%.
47,хуу – синдром двойного игрек (трисомия)
1 на 700 новорожденных. Впервые в 1977году были исследованы.
Высокие мужчины, агрессивное поведение, интеллект снижен или находится на нижней границе нормы. Характерные преступления – поджоги, воровство, детоубийство без мотивации. В больницах закрытого типа, в колонии – 5% таких людей. Поведение детерминировано лишней хромосомой.
47,ххх – синдром Сверхженщины.
1на 1000 новорожденных девочек.
Внешне не проявляется, легкое слабоумие. Считают, что около 1% девушек и женщин с легким слабоумием. Могут беременеть и рождают нормальных детей (во время мейоза происходит самокоррекция).
45,у0 – нежизнеспособны – аборт.
45,х0 синдром Шеришевкого-Тернера
частота встречаемости 1:2000 девочек. Летальность при моносомии очень высокая, каждый 13 выкидыш имеет такую природу. Фенотипические проявления – маленький рост, для многих характерна шейная складка. Локтевой изгиб под углом, укорочены 4 и 5 пальцы, антимонголоидные глаза, абстрактное мышление отсутствует, упорные, трудолюбивые, способны заканчивать школы, ВУЗы. Любовь к опеканию маленьких детей. Отсутствует критическое восприятие своих дефектов. Низкий рост девочки – непременное условие для проведения кариотипирования. Окружность головы больше нормы, груди широко расставлены.
49,ххххх – нарушения те же, Но встречаемость ниже
49,хххху – то же.
Аутосом меньше 44 не бывает, но больше – возможно.
47,хх+21, 47,ху+21 Синдром Дауна.
Частота встречаемости 1на 650 новорожденных.
Фенотипических признаков очень много. Большой язык. Не помещающийся в полости рта, специфический разрез глаз, умственная отсталость и т. д. 12% умственно отсталых детей - Дауны. Частота встречаемости у девочек и мальчиков разных рас примерно одинакова. Чем старше мать, тем выше вероятность рождения ребенка с этой патологией. Каждый 40 ребенок после 40 лет. Не способны к трудовой деятельности, требуют ухода и дорогостоящего лечения.
47,хх+13,47,ху+13 Синдром Патау.
1 больной на 7-8 тысяч новорожденных. Новорожденные имеют нормальные вес и рост. Характерны микроцефалия (недоразвитие головного мозга), резкая умственная отсталость, незарощение неба и губы. Полидактилия, повышенная гибкость суставов, недоразвитие глазного яблока, неправильно сформированные, низко посаженные ушные раковины, пороки внутренних органов. Такие дети не живут долго.
47,хх+18, 47,ху+18 Синдром Эдвардса.
Частота встречаемости у девочек в 3 раза выше, чем у мальчиков.
1 больной на 6-7 тысяч новорожденных.
Характерны множественные аномалии, грубые пороки, характерна грубая задержка роста (гипоплазия в эмбриональном периоде), своеобразный свод черепа, пяткообразно нависающий затылок, короткая шея, расстояние между висками маленькое, ушная раковина деформирована, у половины на затылке избыточная кожа. Продолжительность жизни таких детей снижена. 10% погибают до 1 месяца, 19=0% - до 3 и 30% погибают до года.
Трисомии могут быть по любой хромосоме. Большей частью по 1 паре аутосом. Чем больше генетического материала, тем хуже. В первую очередь страдает интеллект.
Клеточный мозаицизм (генетический) – в соматических клетках одного и того же организма имеется разный набор хромосом. Возникает в результате нерасхождения хромосом во время митоза. По наследству не передается. Проявление зависит от соотношения клеток.
Структурные аномалии хромосом.
Изохромосомы – разделение хромосомы неправильным путем. Чем больше возраст отца, тем, чаще встречается подобное нарушение.
46,хх,5р – дилеция плеча5 хромосомы. Синдром Кошачий крик.
Широко расставленные глаза, физическое недоразвитие. Множественные пороки развития, недоразвита гортань – специфический крик.
Транслокация – обмен участками хромосом (3 вида).
Реципроксные (обмен участками между негомологичными хромосомами).
46,ху, t(9,22) – миелолейкоз (рак крови).
Нереципроксные (между 2мя гомологичными хромосомами). Может не проявляться.
Робертсоновские: возникают при нарушениях деления акроцентрических хромосом. Разрыв по центромере, короткие части дегенерируют, длинные срастаются часто по 15 хромосоме.
46,хх,15t – рак крови. Приводит к ожирению, гипотонии мышц, умственной отсталости. Возможно рождение ребенка – Дауна(5-10% перенос с 21 на 14).
Инверсия – поворот. Кольцевые хромосомы могут возникать по 16и 18 хромосомам, терминальные концы разрываются. Обозначается – Г. По 18 хромосоме – слабоумие, аномалии лица.
В результате хромосомных мутаций и аббераций возникает дисбаланс генетического материала, что приводит к психическим и физическим нарушениям развития. Аномалии по крупным хромосомам происходят значительно реже, чем по мелким. Самая маленькая хромосома – 21, нарушения ее строения встречаются чаще всего. Нехватка генетического материала переносится хуже, чем избыток. Если много эухроматина – нежизнеспособность ребенка, если преобладает гетерохроматин – тяжелые патологии (8,13,18,21,х хромосомы).
Геномные мутации, причины и механизмы их возникновения. Классификация и значение геномных мутаций. Геномные болезни человека. Примеры.
Геномные мутации. Полиплоидия – увеличение числа хромосом, кратное диплоидному набору (клетки печени в норме). Анеуплоидия (гетероплоидия)- уменьшение или увеличение количества хромосом не кратное диплоидному. Гаплоидия – наличие гаплоидного набора хромосом в некоторых клетках (как правило, происходит гибель клеток).
Мутации могут быть полезными, вредными или не оказывать явного влияния – т. е. быть нейтральными. Обычные гены в популяции адаптивны, обладатели лучше приспосабливаются, а вновь возникающие мутации чаще всего уже встречались ранее и были утрачены, потому что не способствовали лучшему приспособлению к определенным условиям жизни. Мутантный ген может накапливаться, может быть полезным. И все же большинство мутаций – вредны.
При геномных мутациях у организма-мутанта происходит внезапное изменение числа хромосом, кратное целому геному. Если через 2n обозначить число хромосом в исходном диплоидном геноме, то в результате геномной мутации, называемой полиплоидизадией, происходит образование полиплоидных организмов, геном которых представлен 4n, 6n и т. д. хромосомами. Различают аллополиплоидию, в результате которой происходит объединение при гибридизации целых неродственных геномов, и аутополиплоидию, для которой характерно адекватное увеличение числа хромосом собственного генома, кратное 2n.
Геномные болезни:
Нерасхождение хромосом при митозе или мейозе
Утрата хромосомы в анафазе полиплоидия
Болезни человека с наследственной предрасположенностью, механизмы их возникновения и проявления. Примеры.
Эта группа болезней отличается от генных болезней тем, что для своего проявления нуждается в действии факторов внешней среды. Среди них также различают моногенные, при которых наследственная предрасположенность обусловлена одним патологически измененным геном, и полигенные. Последние определяются многими генами, которые в нормальном состоянии, но при определенном взаимодействии между собой и с факторами среды создают предрасположение к появлению заболевания. Они называются мультифакториальными заболеваниями (МФЗ).
Заболевания моногенные с наследственным предрасположением относительно немногочисленны. К ним применим метод менделевского генетического анализа. Учитывая важную роль среды в их проявлении, они рассматриваются как наследственно обусловленные патологические реакции на действие различных внешних факторов (лекарственных препаратов, пищевых добавок, физических и биологических агентов), в основе которых лежит наследственная недостаточность некоторых ферментов.
К таким реакциям могут быть отнесены наследственно обусловленная непереносимость сульфаниламидных препаратов, проявляющаяся в гемолизе эритроцитов, повышении температуры при применении общих анестезирующих средств.
Наряду с химическими агентами у людей отмечается наследуемая патологическая реакция на физические факторы (тепло, холод, солнечный свет) и факторы биологической природы (вирусные, бактериальные, грибковые инфекции, вакцины). Иногда отмечается наследственная устойчивость к действию биологических агентов. Например гетерозиготы HbA HbS устойчивы к заражению возбудителем тропической малярии.
К болезням с наследственной предрасположенностью, обусловленной многими генетическими и средовыми факторами, относятся такие заболевания, как псориаз, сахарный диабет, шизофрения. Этим заболеваниям присущ семейный характер, и участие наследственных факторов в их возникновении не вызывает сомнений. Однако генетическая природа предрасположенности к ним пока не расшифрована.
Нередко предрасположенность к ряду заболеваний наблюдается у людей с определенным сочетанием различных генов. Так, у людей со II (А) группой крови чаще наблюдается рак желудка и кишечника, матки, яичников и молочной железы, а также пернициозная анемия, сахарный диабет, ишемическая болезнь сердца, холецистит, желчно-каменная болезнь, ревматизм. У людей с I (0) группой крови чаще встречается язвенная болезнь желудка и двенадцатиперстной кишки. Установление с помощью различных методов генетических исследований точного диагноза заболевания, выяснение роли наследственности и среды в его развитии, определение типа наследования в случае наследственных болезней дают возможность врачу разрабатывать методы лечения и профилактики появления этих заболеваний в следующих поколениях.
^ Другие особенности:
1.Мало различается риск рождения больного ребенка для потомства и сибсов пробанда. Например, расщелина губ и неба составляет для обоих категорий 4%
2.Конкордантность МБ не достигает 100%. как при моногенных, но выше чем показатель дизиготных У МБ 21-63%. Если этот показатель в 4 раза выше. чем у дизиготных –модель полигенного наследования предпочтительнее
3.Риск рождения ребенка с мультифакториальным заболеванием прямо пропорционален тяжести порока:
4.Риск МП для родственников первой степени родства значительно превышает популяционный и выше аналогичного 2 и 3 степени родства. Шизофрения риск для 1 –9-13, а для 2 –3%
5.Риск зависит от числа больных в семье %:чем больше больных. тем больше риск. Например, анэнцефалия после рождения одного больного –2-5%, а после третьего - уже 15-20%
6. Влияние пола
Вывих бедра чаще встречается у девочек. Если пробанд женского пола, то для братьев риск =1%. а для сестер –5%. Если пробанд мужского пола, то для братьев-5%. а для сестер –7%. т. е. порог проявления для чаще поражаемого пола ниже, чем для реже поражаемого.
27.Генеалогический метод изучения генетики человека. Особенности наследования признаков в родословных с аутосомно-доминантным, аутосомно-рецессивным, Х-сцепленным и У-сцепленным типах наследования.
В основе этого метода лежит составление и анализ родословных. Родословные человека составлялись на протяжении многих столетий в отношении царствующих семейств в Европе и Азии.
Как метод изучения генетики человека генеалогический метод стали применять только с начала XX столетия, когда выяснилось, что анализ родословных, в которых прослеживается передача из поколения в поколение какого-то признака (заболевания), может заменить собой фактически неприменимый в отношении человека гибридологический метод.
При составлении родословных исходным является человек — пробанд, родословную которого изучают. Обычно это или больной, или носитель определенного признака, наследование которого необходимо изучить.
С помощью генеалогического метода может быть установлена наследственная обусловленность изучаемого признака, а также тип его наследования (аутосомно-доминантный, аутосомно-рецессивный, X-сцепленный доминантный или рецессивный, Y-сцепленный). При анализе родословных по нескольким признакам может быть выявлен сцепленный характер их наследования, что используют при составлении хромосомных карт. Этот метод позволяет изучать интенсивность мутационного процесса, оценить экспрессивность и пенетрантность аллеля. Он широко используется в медико-генетическом консультировании для прогнозирования потомства. Однако необходимо отметить, что генеалогический анализ существенно осложняется при малодетности семей.
Родословные при аутосомно-доминантном наследовании.
- равная вероятность встречаемости данного признака как у мужчин, так и у женщин. Это обусловлено одинаковой двойной дозой генов, расположенных в аутосомах у всех представителей вида и получаемых от обоих родителей, и зависимостью развивающегося признака от характера взаимодействия аллельных генов.
При доминировании признака в потомстве родительской пары, где хотя бы один родитель является его носителем, он проявляется с большей или меньшей вероятностью в зависимости от генетической конституции родителей (рис. 6.25).
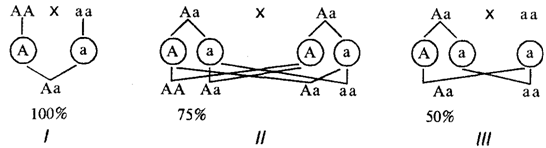
Если анализируется признак, не влияющий на жизнеспособность организма, то носители доминантного признака могут быть как гомо-, так и гетерозиготами. В случае доминантного наследования какого-то патологического признака (заболевания) гомозиготы, как правило, нежизнеспособны, а носители этого признака — гетерозиготы.
Таким образом, при аутосомно-доминантном наследовании признак может встречаться в равной мере у мужчин и у женщин и прослеживается при достаточном по численности потомстве в каждом поколении по вертикали. Анализируя родословные, необходимо помнить о возможности неполного пенетрирования доминантного аллеля, обусловленной взаимодействием генов или факторами среды. Показатель пенетрантности может быть вычислен как отношение фактического числа носителей признака к числу ожидаемых носителей этого признака в данной семье. Необходимо также помнить, что некоторые заболевания проявляются не сразу с момента рождения ребенка. Многие болезни, наследуемые по доминантному типу, развиваются лишь в определенном возрасте. Так, хорея Гентингтона клинически проявляется к 35—40 годам, поздно проявляется и поликистоз почек. Поэтому при прогнозировании подобных заболеваний в расчет не принимаются братья и сестры, не достигшие критического возраста.
Родословные при аутосомно-рецессивном наследовании. Рецессивные признаки проявляются фенотипически лишь у гомозигот по рецессивным аллелям. Эти признаки, как правило, обнаруживаются у потомков фенотипически нормальных родителей — носителей рецессивных аллелей. Вероятность появления рецессивного потомства в этом случае равна 25%. Если один из родителей имеет рецессивный признак, то вероятность проявления его в потомстве будет зависеть от генотипа другого родителя. У рецессивных родителей все потомство унаследует соответствующий рецессивный признак.
Для родословных при аутосомно-рецессивном типе наследования характерно, что признак проявляется далеко не в каждом поколении. Чаще всего рецессивное потомство появляется у родителей с доминантным признаком, причем вероятность появления такого потомства возрастает в близкородственных браках, где оба родителя могут являться носителями одного и того же рецессивного аллеля, полученного от общего предка.
Родословные при доминантном Х-сцепленном наследовании признака. Гены, расположенные в Х-хромосоме и не имеющие аллелей в Y-хромосоме, представлены в генотипах мужчин и женщин в разных дозах. Женщина получает две свои Х-хромосомы и соответствующие гены как от отца, так и от матери, а мужчина наследует свою единственную Х-хромосому только от матери. Развитие соответствующего признака у мужчин определяется единственным аллелем, присутствующим в его генотипе, а у женщин он является результатом взаимодействия двух аллельных генов. В связи с этим признаки, наследуемые по Х-сцепленному типу, встречаются в популяции с разной вероятностью у мужского и женского пола.
При доминантном Х-сцепленном наследовании признак чаще встречается у женщин в связи с большей возможностью получения ими соответствующего аллеля либо от отца, либо от матери. Мужчины могут наследовать этот признак только от матери. Женщины с доминантным признаком передают его в равной степени дочерям и сыновьям, а мужчины — только дочерям. Сыновья никогда не наследуют от отцов доминантного Х-сцепленного признака.
Примером такого типа наследования служит описанная в 1925 г. родословная с фолликулярным кератозом —кожным заболеванием, сопровождающимся потерей ресниц, бровей, волос на голове (рис. 6.30). Характерным является более тяжелое течение заболевания у гемизиготных мужчин, чем у женщин, которые чаще всего являются гетерозиготами.
При некоторых заболеваниях наблюдается гибель мужчин-гемизигот на ранних стадиях онтогенеза. Тогда в родословных среди пораженных должны быть только женщины, в потомстве которых отношение пораженных дочерей, здоровых дочерей и здоровых сыновей равно 1:1:1. Мужские доминантные гемизиготы, не погибающие на очень ранних стадиях развития, обнаруживаются в самопроизвольных абортах или среди мертворожденных. Такими особенностями наследования у человека характеризуется пигментный дерматоз.
Родословные при рецессивном Х-сцепленном наследовании признаков. Характерной особенностью родословных при данном типе наследования является преимущественное проявление признака у гемизиготных мужчин, которые наследуют его от матерей с доминантным фенотипом, являющихся носительницами рецессивного аллеля. Как правило, признак наследуется мужчинами через поколение от деда по материнской линии к внуку. У женщин он проявляется лишь в гомозиготном состоянии, вероятность чего возрастает при близкородственных браках.
Наиболее известным примером рецессивного Х-сцепленного наследования является гемофилия.
Другим примером наследования по данному типу является дальтонизм — определенная форма нарушения цветоощущения.
Родословные при Y-сцепленном наследовании. Наличие Y-хромосомы только у представителей мужского пола объясняет особенности Y-сцепленного, или голандриче-ского, наследования признака, который обнаруживается лишь у мужчин и передается по мужской линии из поколения в поколение от отца к сыну.
Одним из признаков, Y-сцепленное наследование которого у человека все еще обсуждается, является гипертрихоз ушной раковины, или наличие волос на внешнем крае ушной раковины. Предполагают, что в коротком плече Y-хромосомы кроме этого гена находятся гены, определяющие мужской пол. В 1955 г. у мыши описан определяемый Y-хромосомой трансплантационный антиген, названный HY. Возможно, он является одним из факторов половой дифференцировки мужских гонад, клетки которых имеют рецепторы, связывающие этот антиген. Связанный с рецептором антиген активизирует развитие гонады по мужскому типу (см. разд. 3.6.5.2; 6.1.2). Этот антиген в процессе эволюции остался почти неизменным и встречается в организме многих видов животных, в том числе и человека. Таким образом, наследование способности к развитию гонад по мужскому типу опред