Поясните понятия: что такое плюрипотентность и полипотентность, детерминация, морфогенез, гомеозисные гены? Каково значение гибели клеток в эмбриогенезе.
Плюрипотентность — способность к дифференцировке разных тканей разных зародышевых листков.
Полипотентность — способность генома стволовых клеток взрослого организма изменять профиль дифференцировки при трансплантации в новую ткань реципиента.
Детерминация - определение пути развития. Клеточная система выбирает один из многих путей развития.
Морфогенез -формирование пространственной организации организма и его частей. Клетка «знает» свое место и дифференцируется в соответствии со своим положением Создается морфогенетическое поле-зона, в пределах которой клетки обмениваются сигналами позиционной информации и дифференцируются, помня о своем исходном назначении.В ходе морфогенеза происходят: пролиферация, межклеточное взаимодействие, индукция, миграция, гибель клеток.
Гомеозисные гены детерминируют процессы роста и дифференцировки. Мутации в гомеозисных генах могут вызвать превращение одной части тела в другую. Гомеозисными мутантами называются такие организмы, у которых на месте органа развивается орган другого типа. Гомеозисные гены контролируют работу других генов и определяют превращение внешне неразличимых участков зародыша или определённого органа (ткани, участка тела).
Апоптоз - программированная клеточная смерть. Естественный процесс массовой гибели клеток в ходе эмбрионального развития, гистогенеза и морфогенеза. Например пальцы конечностей формируются при условии апоптоза.Организмы с обширными дефектами, обусловленными нарушениями апоптоза, погибают ещё на ранних стадиях онтогенеза. Патологические процессы развиваются в случае подавления или усиления апоптоза. При недостаточности апоптоза прогрессируют аутоиммунные процессы и злокачественные новообразования. При усилении апоптоза возникают аплазии и дегенеративные процессы, а также некоторые уродства с дефектами тканей.
Периоды пренатального развития,их продолжительность.
1.Начальный(концептус).
Критические события- оплодотворение.
Продолжтельность-2 недели
2.Зародышевый(эмбрион)
Критические события- образование первичной полоски
Продолжительность-от 3-й по 8-ю недели включительно
3.Плодный(плод)
Критические события- плацентация
Продолжительность-до конца беременности.
Мейоз.
Мейоз- редукционное деление клетки — деление ядра эукариотической клетки с уменьшением числа хромосом в два раза.
Мейоз состоит из 2 последовательных делений с короткой интерфазой между ними.
Профаза I — профаза первого деления очень сложная и состоит из 5 стадий:
1) Лептотена или лептонема — упаковка хромосом, конденсация ДНК с образованием хромосом в виде тонких нитей (хромосомы укорачиваются). 2)Зиготена или зигонема — происходит конъюгация — соединение гомологичных хромосом с образованием структур, состоящих из двух соединённых хромосом, называемых тетрадами или бивалентами и их дальнейшая компактизация. 3)Пахитена или пахинема — (самая длительная стадия) кроссинговер (перекрест), обмен участками между гомологичными хромосомами; гомологичные хромосомы остаются соединенными между собой. 4)Диплотена или диплонема — происходит частичная деконденсация хромосом, при этом часть генома может работать, происходят процессы транскрипции (образование РНК), трансляции (синтез белка); гомологичные хромосомы остаются соединёнными между собой. У некоторых животных в ооцитах хромосомы на этой стадии профазы мейоза приобретают характерную форму хромосом типа ламповых щёток. 5)Диакинез — ДНК снова максимально конденсируется, синтетические процессы прекращаются, растворяется ядерная оболочка; центриоли расходятся к полюсам; гомологичные хромосомы остаются соединёнными между собой.
К концу Профазы I центриоли мигрируют к полюсам клетки, формируются нити веретена деления, разрушаются ядерная мембрана и ядрышки
Метафаза I — бивалентные хромосомы выстраиваются вдоль экватора клетки. Анафаза I — микротрубочки сокращаются, биваленты делятся и хромосомы расходятся к полюсам. Важно отметить, что, из-за конъюгации хромосом в зиготене, к полюсам расходятся целые хромосомы, состоящие из двух хроматид каждая, а не отдельные хроматиды, как в митозе. Телофаза I — хромосомы деспирализуются и появляется ядерная оболочка.
Второе деление мейоза следует непосредственно за первым, без выраженной интерфазы: S-период отсутствует, поскольку перед вторым делением не происходит репликации ДНК.
Профаза II — происходит конденсация хромосом, клеточный центр делится и продукты его деления расходятся к полюсам ядра, разрушается ядерная оболочка, образуется веретено деления. Метафаза II — унивалентные хромосомы (состоящие из двух хроматид каждая) располагаются на «экваторе» (на равном расстоянии от «полюсов» ядра) в одной плоскости, образуя так называемую метафазную пластинку.
Анафаза II — униваленты делятся и хроматиды расходятся к полюсам. Телофаза II — хромосомы деспирализуются и появляется ядерная оболочка.
Альбина Идрисова
Мальформации и их причины
1.Дефекты гомейозисных генов/морфогенов:
Примеры: дефекты гена комплекса hox7 связаны с возникновением врожденной расщелины неба; дефект гена shh явл. причиной нарушения дифференцировки переднего мозга на полушария, эта аномалия может сочетаться с недоразвитием носа и верхней губы, отсутствие обонятельного нерва и мозолистого тела.
Дизрупции и тератогены.
Дизрупции (разрушения)
Под дезрупциями понимают аномалии развития плода, которые возникают вследствие воздействия повреждающих факторов, которые нарушают развитие и дифференцировку тканей ребенка. Причиной могут стать радиация, инфекция, воздействие тератогенов и ядов, амниотические перетяжки, механические причины, сосудистый фактор (нарушение кровообращения в бассейне какой-либо артерии). Дизрупции могут возникать в эмбриональном периоде, и тогда они фенотипически выглядят как классические пороки развития, например, анэнцефалия, незаращение губы и неба, пороки развития конечностей по редукционному типу. Большая часть дезрупций всё же возникает уже в плодовом периоде.
Самая сложная дифференциальная диагностика между пороками развития и дизрупциями по сосудистой причине:
При разрушении капиллярной сети эмбриона возможно возникновение таких тяжелых аномалий, как редукционные аномалии конечностей, гипоплазия нижнечелюстной области и конечности.
При сохранении эмбриональных сосудов, которые в норме редуцируются, возникает лучевая косорукость, тибиальная аплазия, косолапость.
При преждевременной ампутации эмбриональных сосудов возникают такие аномалии как гастрошизис, болезнь Клиппеля-Фейля, Мебиуса, Поланда, подковообразная почка.
При нарушенном созревании сосудов возникают капиллярные гемангиомы, артерио-венозные свищи, аневризма Бери.
Окклюзия сосудов вследствие их эмболии вызывает пороки развития головного мозга, атрезия желчного пузыря, анорхизм, аплазии кожи.
Воздействие некоторых тератогенов:
Алкоголь - задержка физического развития. Необычный фенотип (короткие глазные щели). Микроцефалия, умственная отсталость
Тетрациклин - врождённая гипоплазия эмали зубов. Окрашенность зубов (жёлтые зубы)
Ретинол - спонтанные аборты, Черепно-лицевые аномалии. Дефекты невральной трубки
Курение - спонтанные аборты, Внутриутробная задержка роста
Сахарный диабет у матери - врождённые пороки сердца. Синдром каудальной регрессии
45. Критические периоды возникновения врожденны пороков и частота выявляемых аномалий.
На 1000 живых новорожденных:
ЦНС (3-16неделя) – 10 случаев
Сердце(3 – 6 неделя) – 8 случаев
Почки(5 – 8 неделя) - 4 случая
Конечности(4 – 5 неделя) - 2 случая
Верхняя губа(5 – 6 неделя)
Небо(7 – 8 неделя)
Половые органы(7 – 9 неделя) - 6 случаев
Всего 30 случаев на 1000 живорожденных
Алёна
Сиреномелия
Данное заболевание является врожденной патологией, которая возникает вследствие нарушения кровоснабжения и характеризуется слиянием нижних конечностей в сочетании с агенезией почек, аплазией крестца, прямой кишки и мочевого пузыря. Ранее считалось, что эта патология представляет собой тяжелую форму синдрома каудальной регрессии.
При этом Часто отмечаются пороки сердца, почек, передней брюшной стенки, грудной клетки и нижних отделов позвоночника. Также часто выявляются единственная артерия пуповины, неперфорированный анус и агенезия половых органов.
Порок может касаться мягких тканей и некоторых трубчатых костей. Наряду с этим наблюдается гипоплазия или аплазия костей конечностей и таза, врожденный вывих бедра, сгибательные контрактуры тазобедренных и коленных суставов, косолапость. Могут быть сформированы две стопы,одна или отсутствовать.
Синдактилия и полидактилия.
Синдактилия
врожденное полное или неполное сращение пальцев кисти стопы в результате не наступившего их разъединения в процессе эмбрионального развития. Встречается одинаково часто у мужчин и женщин. Односторонняя С. отмечается примерно 2 раза чаще двусторонней. Нередко сочетается с другими пороками развития (Пороки развития). Различают простую и сложную, полную и неполную формы С. Простая форма включает кожную, многослойную и костную С.; сложная — кожную, многослойную, костную и сочетанную. Чаще обнаруживается сращение III и IV пальцев, реже III—IV—V, II—III и IV пальцев. Возможно сращение нескольких пальцев в единый конгломерат, при этом нередко имеются амниотические перетяжки. В большинстве случаев С. пальцы недоразвиты и деформированы.
Полидактилия
врожденная аномалия, представляющая собой увеличение количества пальцев на конечностях. Чаще всего встречается добавочный VI палец на одной или обоих кистях либо на стопах. Но количество их может быть и значительно большим. Добавочные пальцы могут быть сформированы достаточно хорошо и правильно, но чаще встречаются в виде рудиментов (небольшие деформированные части пальцев, свисающие на тонкой кожной ножке). Иногда добавочные пальцы срастаются с V пальцем (синдактилия).
Айнур
51.Эмбриопатия - патология эмбриона, возникающая между 16-м и 75-м днем беременности, индуцированная воздействием повреждающих факторов. Эмбриопатии характеризуются нарушениями формирования органов, которые в конечном счете заканчиваются или гибелью эмбриона, или врожденными пороками развития. В настоящее время причины, вызывающие большинство пороков развития, неизвестны.
К важнейшим порокам развития относятся врожденные пороки сердца, центральной нервной системы, органов пищеварения, пороки развития почек, врожденные пороки лица.
Фетопатии — заболевания плода. Различают ранние фетопатии (возникают в раннем фетальном периоде эмбриогенеза — до 28-недельного срока беременности) и поздние фетопатии (после 28 недель беременности и до начала родов). Причины, вызывающие ранние фетопатии, действуют на плод преим, гематогенно через плаценту. К ранним инфекционным фетопатиям относят: листериоз, орнитоз, цитомегалию, токсоплазмоз, сывороточный гепатит, врождённые респираторные вирусные инфекции фетальный сепсис, фетальную пневмонию различной этиологии, лептоспироз.
Агенезия - (agenesis) - врожденное отсутствие какого-либо органа, обычно связано с пороком или недоразвитием эмбриона.
Аплазия (aplasia; а- + греч. plasis формирование, образование; син. агенезия) - общее название аномалий развития, при которых отсутствует часть тела, орган или его часть, участок какой-либо ткани.
Гипоплазия - это аномалии развития, заключающиеся в недоразвитии ткани, органа, части тела или целого организма.
Гиперплазия (новолат. hyperplasia; др.-греч. ὑπερ- — сверх- + πλάσις — образование, формирование) — увеличение числа структурных элементов тканей путём их избыточного новообразования.
Атрезия — врождённое отсутствие или приобретенное заращение естественных отверстий и каналов в организме. В большинстве случаев атрезия имеет характер врождённой аномалии, реже является следствием иных патологических процессов.
Стеноз (греч. stenosis — сужение, стеснение, от stenos — узкий, тесный), сужение физиологического отверстия (например, левого предсердно-желудочкового отверстия сердца
Эктопия (др.-греч. ἔκτοπος — «смещенный») — нарушение развития в медицине и биологии. Смещение органа или ткани в необычное место, часто в соседние полости тела или наружу.
Персистенция (лат. persisto — постоянно пребывать, оставаться) — способность глаза, (эффект) запоминать последовательные события. Этот эффект основан на инерции человеческого глаза.
диссинхрония - это состояние интеллектуально одаренного ребенка, испытывающего затруднения при контактах со своим окружением.
Амелия (Amelia) - врожденное отсутствие рук или ног вследствие дефектов внутриутробного развити
Фокомелия (от греч. phoke – тюлень и mélos – часть тела), врождённое недоразвитие всех или некоторых конечностей, при котором хорошо развитые кисти и стопы (или голени и предплечья) начинаются непосредственно от туловища, напоминая ласты тюленя.
ЭКТРОМЕЛИЯ (ectromelia) врожденное отсутствие или укорочение (аплазия) длинных костей одной или нескольких конечностей.
52.Буллёзный пемфигоид — доброкачественное хроническое заболевание кожи; первичный элемент — пузырь, формирующийся субэпидермально без признаков акантолиз
Причины и патогенез пемфигоида буллезного
В последние годы исследованиями доказано, что в патогенезе дерматоза важную роль играют аутоиммунные процессы. У больных буллезным пемфигоидом в сыворотке крови и пузырной жидкости обнаружены LgG-антитела, lgA-антитела к базальной мембране, отложение IgG, реже - IgA и СЗ-компонента комплемента в области базальной мембраны как кожи, так и слизистой оболочки. Установлено, что титр антител и циркулирующих иммунных комплексов при пемфигоиде коррелирует с активностью болезн
53.Под названием синдром Элерса — Данло объединяют группу наследственных аномалий с повышенной подвижностью суставов и кожными проявлениями
54.Синдром Марфана (Болезнь Марфана, Marfan syndrome) — заболевание из группы наследственных коллагенопатий, заболеваний соединительной ткани человека. Частный случай дифференцированной дисплазии соединительной ткани человека.
55Синдром Вильямса (синдром «лица эльфа») — синдром, возникающий как следствие хромосомной патологии, страдающие которым обладают специфической внешностью и характеризуются общей задержкой умственного развития при развитости некоторых областей интеллекта.
Маша К.
Несовершенный остеогенез.
Это гетерогенная группа наследственных заболеваний, характеризующихся повышенной ломкостью костей, вследствие нарушения остеогенеза («хрустальные дети»).
Люди с НО либо имеют недостаточное количество коллагена, либо его качество не соответствует норме. Так как коллаген важный белок в структуре кости, это заболевание влечёт за собой слабые или ломкие кости.
Будучи генетическим нарушением, НО является аутосомно-доминантным дефектом. В большинстве передается по наследству от родителей. Представляет редкое заболевание соединительной и опорной ткани с частотой проявления 1:10 000 - 1:20 000 новорожденных. В основе заболевания лежат мутации в одном из двух генов, кодирующих коллаген тип I.
Черепно-ключичный дизостоз.
Относится к смешанным формам системных заболеваний скелета. Этиология и патогенез неизвестны. Наследуется по аутосомно-доминантному типу. Наиболее характерными клинико-рентгенологическими проявлениями черепно-ключичного дизостоза являются специфические врожденные изменения ключиц: обычно отсутствует акромиальный отдел или ключица фрагментирована, лопатки уменьшены в размерах. Изменён череп: увеличена мозговая и уменьшена лицевая части. Наблюдаются изменения свода черепа. Верхняя челюсть с придаточными полостями недоразвиты, уменьшены в объёме, твёрдое нёбо укорочено. Характерно запоздалое и несовершенное развитие постоянных зубов. Иногда не срастаются дужки позвонков. При молекулярно-генетическом исследовании обнаруживаются мутации гена RUNX2 у 60% -70% людей с диагнозом ключично-черепной дизостоз.
59. Аномалии формирования черепа (скафоцефалия, плагиоцефалия) и примеры синдромов с дефектами черепа.
Скафоцефалия возникает при саггитальном краниосиностозе – преждевременном заращении саггитального шва черепа, который начинается от лобной кости и заканчивается в области затылка. Другое название этой деформации головы – ладьевидный череп. Частота аномалии среди всех изолированных краниосиностозов достигает 60%, встречается наиболее часто. При этой деформации череп не может расти в ширину, и увеличивается в переднезаднем направлении, височные области оказываются вдавленными, а лобные и затылочные напротив, нависают. Лицо становится узким, овальным.
Плагиоцефалия - любое искажение или нарушение формы головы, возникающее обычно в результате неправильного закрытия швов между костями черепа (скошенность или несимметричность черепа). Коронарный шов расположен поперек черепа, отделяет лобную кость от остальных костей черепа. При закрытии одной его половины раньше времени формируется плагиоцефалия, своеобразная не симметричная деформация. При этом у ребенка имеется уплощение лобной кости и надбровья с одной стороны, со второй стороны половина лба компенсаторно нависает. По мере роста в деформацию вовлекаются другие лицевые кости, становится всё более плоской скуловая область, нос искривляется в здоровую сторону. Характерно косоглазие.
Пороки развития свода и основания черепа:
а) краниосиностозы (тригоноцефалия, скафоцефалия, плагицефалия, двусторонний коронарный синостоз, и т.д.)
б) черепно-ключичный дизостоз.
Аномалии черепа вследствии раннего зарастания швов: синдром Пфейффера, синдром Крузона, ахондроплазия.
Коагулопатии
собирательное обозначение болезненных состояний, обусловленных нарушениями физиологических механизмов свертывания крови, приводящие обычно к развитию геморрагического синдрома.
Наследственные коагулопатии вызываются генетически обусловленным снижением или извращением плазменных компонентов гемостаза. Наиболее распространенными формами являются гемофилия А, В, С, афибриногенемия.
Наследственные коагулопатии делятся на:
- Гемофилии: А-дефицит VIII фактора, В-дефицит IX фактора, С-дефицит XI фактора, Д-дефицит XII;
- Парагемофилия: дефицит II, V, VII, X факторов;
- Нарушение образования фибрина, дефицит фибриногена (I фактора).
68.Болезнь фон Виллебранда
наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии. Заболевание наследуется по принципу аутосомного доминирования.
Причина кровотечений — нарушение свертываемости крови из-за недостаточной активности фактора Виллебранда.
Наиболее характерным и специфическим симптомом при болезни Виллебранда являются кровотечения из
слизистых полости рта, носа, внутренних органов.
Надя
Дефекты дуги аорты
Коарктация аорты - это врожденное сегментарное сужение грудной аорты, создающее два режима кровообращения в большом круге и вызывающее определенные клинические симптомы.
Перерыв дуги аорты может быть полным или (реже) сегментарным, когда между дугой и нисходящей аортой выявляется атрезированный сегмент в виде фиброзного тяжа. Данная форма порока может рассматриваться как крайняя степень коарктации.
Аневри́зма — выпячивание стенки артерии (реже — вены) вследствие её истончения или растяжения.
Дуга аорты двойная – дуга аорты представлена двумя стволами: один располагается впереди трахеи, другой – позади пищевода. Слева от них оба ствола соединяются вновь. Передняя дуга обычно тоньше задней или представлена плотной связкой. Образуется двойная дуга в результате персистирования правой IV аортальной дуги.
Дуга аорты правосторонняя – развивается из эмбриональной правой дуги при редукции левой. Располагается позади пищевода. Из остатков левой дуги в таких случаях нередко образуется дивертикул.
Дуга аорты шейная – в случае инволюции 4 жаберных дуг дуга аорты может развиться из артерии III жаберной дуги. В этом случае дуга аорты располагается на шее над вырезкой грудины. Встречается чрезвычайно редко.
Саша- НЕТ ОТВЕТОВ
Алина (PS Алине можно было так оставить с фотографиями, так как она делал воловину из этого сама, надеюсь никто не против)
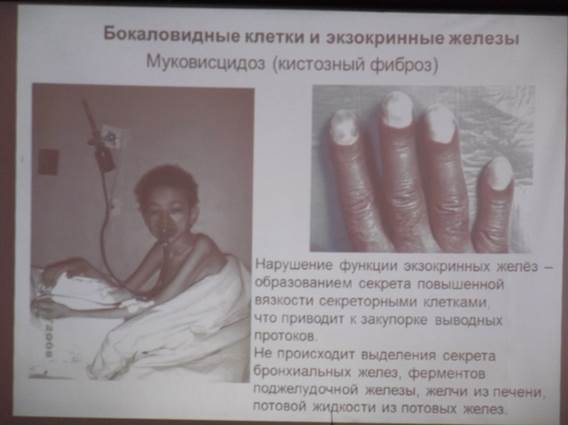
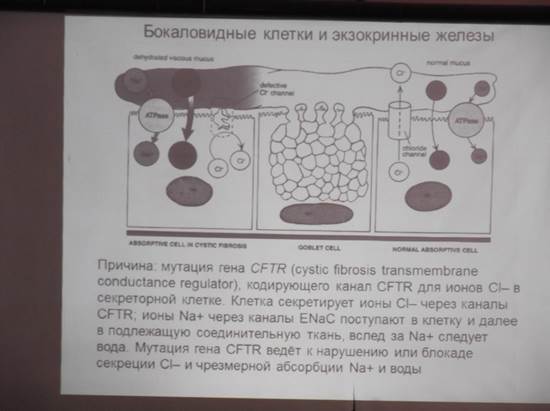
1. Муковисцидоз

2. Атрезия и стеноз желчных протоков
Атрезия желчных путей (особенно внутрипеченочных) в большинстве случаев связана с перенесенным внутриутробно гепатитом, чаще вызванным одним из реовирусов. У некоторых детей возникновение этого порока развития обусловлено неблагоприятными факторами, действовавшими на 4–8-й нед внутриутробной жизни.
В этиологии атрезии желчных протоков большинство отечественных и зарубежных авторов придают значение таким факторам, как продуктивное воспаление, вызывающее дегенерацию эпителия протоков, облитерацию их просвета и околопротоковый склероз. Прогрессирование процессов альтерации, пролиферации и фибро-зирования во внутриутробный период и после рождения ведет к полной обтурации просвета желчных протоков.
Патогенез болезни связан с нарушением выделения желчи вследствие непроходимости желчных протоков. Развиваются желтуха, билиарный цирроз печени, портальная гипертензия, печеночная недостаточность. При отсутствии хирургического лечения дети умирают вскоре после рождения.
Патологическая анатомия. Желчные протоки в месте атрезии представлены тонким фиброзным тяжем.
Стеноз желчных протоков
Заболевание представляет собой комплекс различных нарушений, характеризующихся общим признаком — затруднением оттока желчи из желчных протоков в двенадцатиперстную кишку. Стеноз возникает обычно в нижнем отделе общего желчного протока на почве желчнокаменной болезни, воспалительного или опухолевого процесса в протоке, в сфинктере Одди, ампуле и папилле Фатера, а также вследствие фиброза, рубцевания и спаек.
3. Трахео-пищеводные свищи
Развивающиеся из одного зачатка пищеводная и дыхательная трубки при различных нарушениях в течение внутриутробного периода имеют патологические сообщения между собой — свищи. Наиболее часто свищ возникает между трахеей и пищеводом. Во время кормления у ребенка возникает кашель, он синеет (цианоз). Особенно яркая картина развивается при больших размерах свища. На фоне попадания пищи в дыхательные пути может развиться пневмония.
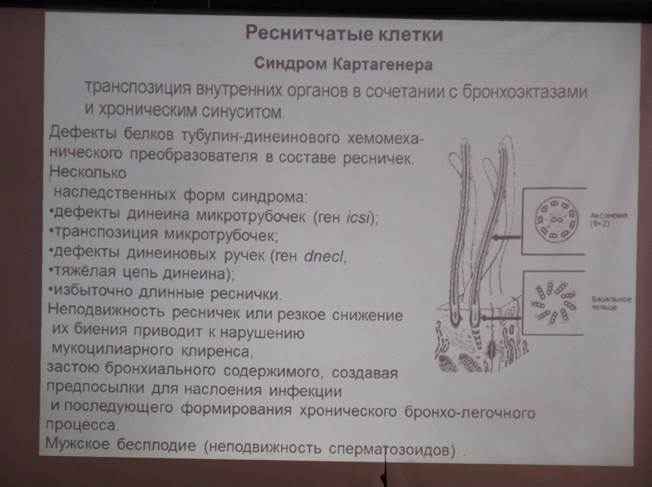
4. Синдром Картагенера
комбинированный порок развития, характеризующийся образованием бронхоэктазов в сочетании с полным или частичным обратным расположением внутренних органов и полипозом слизистой оболочки носа. Часто сочетается с другими врожденными аномалиями: полидактилией, агенезией или гипогенезией лобных пазух, пороками развития позвонков и ребер, мочевыводящих путей, сердца, гипофункцией некоторых эндокринных желез (щитовидной, гипофиза, надпочечников) с задержкой роста, поражением сетчатки (пигментный ретинит, расширение сосудов сетчатки) и др.
Этиология и патогенез. Не изучены. Имеет значение генетическая предрасположенность, передаваемая по аутосомно-рецессивному типу. Не исключена возможность врожденной предрасположенности бронхиального дерева к хроническому воспалению с последующим развитием бронхоэктазов.
5. Респираторный дистресс-синдром новорожденных

Дина
104. Врождённый альвеолярный протеиноз (ЛАП):
Это врожденное нарушение образования и накопления сурфактанта пневмоцитами типа II и альвеолярными макрофагами. Происходит прогрессирующее накопление хлопьевидного сурфактанта, клеточных обломков и пенистых макрофагов в альвеолярном пространстве, возможности для газообмена уменьшаются. Клинически: кашель, гипоксия, отставание в физическом развитии, легочные инфекции.
105. Первичная (врождённая) эмфизема (недостаточность α1-антитрипсина)
Обусловлена недостаточностью α1-антитрипсина. Приводит к развитию панлобулярной (охватывающей все отделы ацинуса) эмфиземы (повышению воздушности легочной ткани, разрушению межальвеолярных перегородок). При воспалительных процессах (например, при курении) моноциты и нейтрофилы высвобождают большое количество ферментов (эластаза, химотрипсин, трипсин). α1-антитрипсин разрушает протеазы, предотвращая повреждение тканей. При недостаточности α1-антитрипсина в легких происходит усиленный протеолиз, что приводит к разрушению межальвеолярных перегородок и эластического каркаса альвеол, расширению респираторных отделов дыхательных путей и снижению газообмена.
Поликистоз почек
Этоврожденное заболевание, при котором в обеих почках появляются и постепенно увеличиваются кисты, что приводит к атрофии функционирующей паренхимы. Относится к наследственным аномалиям развития.
Причина возникновения аномалии неизвестна. Патогенез обусловлен пороком эмбрионального развития канальцев, часть которых трансформируется в кисты. Почки у большинства больных увеличены, содержат множество кист различных размеров, между которыми расположены участки сохранившейся паренхимы, местами замещенной соединительной тканью. Чашечки и лоханки сдавлены и деформированы. Кисты могут нагнаиваться.
На макроуровне для поликистоза характерно наличие множественных кист (отсюда название: поли- + киста + -оз) в обеих почках. Кисты образуются из-за повышенной пролиферации и дедифференциации фильтрующего эпителия нефрона. В результате вместо нормальных почечных канальцев образуются наполненные жидкостью пузырьки — кисты, что приводит к значительному увеличению объема почек (вес почки больного может достигать 35 кг). Кисты в почке больного возникают фокально не более чем в 2-5 % нефронов, но из-за увеличения объёма кист происходит сдавление соседних здоровых нефронов, и постепенно почка теряет фильтрующую функцию.
Экстрофия мочевого пузыря
Это врождённый порок развития мочевого пузыря, при котором мочевой пузырь оказывается не внутри, а снаружи. Передняя стенка мочевого пузыря отсутствует, как и соответствующий ей участок брюшной стенки, которая расщеплена, и таким образом мочевой пузырь оказывается вовне. Моча льётся наружу через отверстия мочеточников.
Состояние экстрофии образуется очень рано во время развития эмбриона, примерно на 4-5 неделях его развития.
Экстрофия мочевого пузыря часто сочетается с пороками развития верхних мочевых путей, а также с аномалиями других органов и систем.
Экстрофированная слизистая стремительно, уже к концу первых суток жизни, подвергается изменениям в связи с постоянным раздражением и присоединением воспаления. Быстро развивается восходящая инфекция мочевых путей. Причины неправильного развития пока не до конца ясны. Именно в это время начинают формироваться органы и ткани организма из разъединяющихся, делящихся и соединяющихся клеток. Одна из теорий предполагает, что именно из-за какого-то неправильного механизма такого деления и соединения клеток, что-то происходит с клоакальной мембраной, что не даёт ей закрыться, и это приводит к тому, что мочевой пузырь оказывается вне брюшной полости. Другая теория предполагает, что на этом этапе развития эмбриона слой клеток над мочевым пузырем очень тонок, он не может удержать мочевой пузырь внутри и расщепляется, опять же мочевой пузырь оказывается снаружи.
Дивертикул Меккеля
Это локальное мешковидное выпячивание стенки подвздовшной кишки, образовавшеяся вследствие неполного заращения желточного протока, который участвует в питании плода(в норме подвергается обратному развитию после 6—8 нед. внутриутробного периода). Может никак себя не проявлять, но может воспаляться и симулировать клиническую картину острого аппендицита.
Наиболее частыми клиническими проявлениями М.д, служат его воспаление, непроходимость кишечника и кишечное кровотечение. Воспаление (дивертикулит) могут спровоцировать инородные тела, гельминты, попавшие в просвет дивертикула, а также застой кишечного содержимого
Тимур
109.Урахус
Урахус— трубчатое образование у эмбриона, соединяющее передний отдел верхушки мочевого пузыря и пупок между брюшиной и поперечной фасцией живота, образуется из верхнего отдела аллантоиса. По данному протоку моча плода выводится в околоплодные воды. С 5 месяцев внутриутробной жизни начинается облитерация протока, которая завершается к моменту рождения, с превращением его в срединную пупочную связку. Однако при определенных условиях проток перекрывается не полностью, в результате чего формируются его аномалии:
-Пупочный свищ — незаращение части урахуса, находящейся в области пупка. Проявляется такая аномалия постоянным мокнутием ранки пупка.
-Пузырно — пупочный свищ — полное незаращение урахуса. Данный вид характеризуется постоянным выделением из ранки пупка мочи.
-Дивертикул мочевого пузыря — незаращение части урахуса, отходящей от мочевого пузыря.
-Киста урахуса — незаращение средней части урахуса.
110. Пузырный занос (полный и неполный) и его причины
Пузырный занос - заболевание плодного яйца, отличительными признаками которого являются перерождение ворсин хориона в пузырьки с жидкостью, разрастание эпителия ворсин, особенно синцития.
Причиной возникновения пузырного заноса является наличие у эмбриона двойного набора хромосом отца при недостаточном количестве или же вообще отсутствии хромосом матери. Такая аномалия случается, когда одновременно 2 сперматозоида оплодотворяют «неполноценную» яйцеклетку - с задержкой набора хромосом или безъядерную. При этом в первом случае развивается неполный пузырный занос, а во втором - полный.
Полный пузырный занос возникает при однородительской дисомии, когда по неизвестным причинам происходит потеря материнских генов и дублирование отцовского гаплоидного генома (зигота имеет кариотип 46,ХХ). Иногда (5%) полный пузырный занос вызван оплодотворением пустой (безъядерной) яйцеклетки двумя сперматозоидами, приводящим к кариотипу 46,XY или 46,XX. Эмбрион погибает на ранних стадиях развития, до установления плацентарного кровообращения
Неполный пузырный занос вызван триплоидией в результате оплодотворения яйцеклетки двумя сперматозоидами (диспермия) с задержкой гаплоидного набора материнских хромосом. Клетки концептуса содержат один гаплоидный набор материнских хромосом и диплоидный набор отцовских хромосом - кариотип может быть 69.XXY, 69.ХХХ или 69.XYY. Плод погибает на 10 нед внутриутробного развития.
Патогенез:
При значительном накоплении жидкости в ворсинах сосуды трофобласта атрофируются. Синцитий, покрывающий пузырьки, способен пролиферировать и ферментативно расплавлять децидуальную оболочку, прорастать и внедряться в мышечный слой матки, разрушая мышечные элементы и сосуды. Иногда инвазивная способность покровного эпителия пузырьков столь значительна, что они разрушают стенку матки, проникают в брюшную полость и могут послужить причиной внутреннего кровотечения. Это деструирующая форма пузырного заноса, которая по характеру роста напоминает опухоль. Обычно она связана с опасным для жизни кровотечением.
111.
Омфалоцеле
Омфалоцеле (грыжа пупочного канатика, пуповинная грыжа, эмбриональная грыжа) — вид врождённого дефекта передней брюшной стенки, при котором петли кишечника, печень и, иногда, другие органы выходят за пределы брюшной полости в грыжевом мешке. Омфалоцеле обусловлено дефектом развития мышц передней брюшной стенки
В некоторых случаях, предположительно, омфалоцеле может являться следствием генетического рассройства (синдром Эдвардса, синдром Патау).
В норме в ходе эмбриогенеза петли кишечника выходят за пределы брюшной полости, выпячиваясь в пупочный канатик, однако на сроке десяти недель беременности возвращаются обратно; при омфалоцеле же органы остаются в пупочном канатике. Грыжевой мешок при омфалоцеле в лёгких случаях может содержать единичные петли кишечника, в тяжёлых — практически все органы живота.
Сочетается с другими аномалиями и хромосомными дефектами в 70% случаев. Хромосомные аномалии встречаются в 20% случаев (синдром Эдвардса, синдром Патау).. Из многочисленных сочетанных аномалий, встречающихся при омфалоцеле, особое внимание занимает синдром Беквита- Видемана (пуповинная грыжа, макроглоссия, органомегалия). Наблюдается у 12% детей с омфалоцеле.
112.Многоводие и маловодие
Околоплодные воды, или амниотическая жидкость (АЖ) — среда обитания плода, выполняющая одновременно несколько функций: создание пространства для свободных движений растущего плода, защита от механической травмы, поддержание температурного баланса, предотвращение компрессии пуповины в родах, осуществление транспортной функции и участие в обмене веществ.
Многоводие – это патология беременности, при которой происходит избыточное накопление околоплодных вод - более 1,5- 2 литра жидкости. Соответственно, маловодие - это недостаток околоплодных вод, при котором количество околоплодной жидкости составляет менее 500 мл.
113. Синдром (последовательность) Поттера
Синдром Поттера представляет собой сочетание двусторонней агенезии почек и характерных аномалий лица (приплюснутый нос, гипертелоризм, эпикант, узкие глазные щели, борозда под нижними веками, микрогнатия и мягкие, большие, деформированные, низко расположенные ушные раковины), 40% детей рождаются недоношенными, большинство погибают в первые часы жизни. При этом синдроме могут встречаться следующие пороки развития: двусторонняя гипоплазия легких, аномалии гениталий, атрезия ануса, отсутствие сигмовидной и прямой кишки, атрезия пищевода и двенадцатиперстной кишки, единственная пупочная артерия и деформация нижней челюсти туловища и нижних конечностей. Типичное лицо, гипоплазия легких и аномалии конечностей развиваются вследствие характерного для данного синдрома маловодия. Описаны случаи односторонней агенезии почек у родственников
Данил
114.Placenta accrete
Отделение зрелой плаценты от стенки матки в III периоде родов в нормальных условиях происходит довольно быстр