течения гепатита вплоть до формирования цирроза, особенно когда исключены наиболее частые его причины, и возраст больного или детский (с 5 лет), или молодой (до 30 лет).
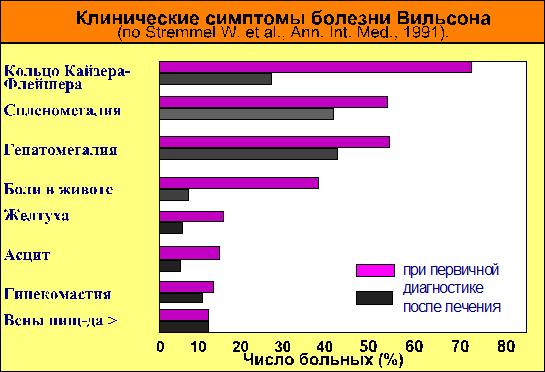
Решающее значение в диагностике болезни Вильсона может иметь обнаружение радужного кольца Кайзера-Флейшера. Наличие его у больного сразу установить диагноз заболевания. Но выявить его удается преимущественно у больных с первичной неврологической манифестацией, только тогда кольцо обнаруживается почти у всех больных. Но на ранних стадиях болезни, например, при первично печеночной форме, и особенно при фульминантном гепатите вследствие болезни Вильсона, кольцо Кайзера-Флейшера часто отсутствует.
При фульминантном течении гепатита у больных дополнительную помощь может оказать обнаружение внепеченочных симптомов. В ряде случаев отмечается голубое окрашивание ложа ногтей и голубоватый оттенок кожи в области больших берцовых костей (118).
Наряду с клиническими данными для установления диагноза используются лабораторные методы, но в педиатрической практике их роль оценивается по-разному.
К одним из основных относится определение в крови уровня церулоплазмина (белков, доставляющих медь в печень). Нормальное значение церулоплазмина в сыворотке крови считается > 25 мг% при использовании нефелометрического метода исследования. Концентрация этого белка в крови при болезни Вильсона уменьшается до 20 мг% и ниже. Уровень церулоплазмина снижен приблизительно у 65% с печеночной формой и до 85% больных с неврологической симптоматикой. Правда, у 20% здоровых гетерозиготных носителей генетического дефекта также обнаруживают пониженные значения этого параметра (118).
У детей с болезнью Вильсона большинство исследователей также отмечает, что уровень этого белка часто снижен. Несмотря на то, что у больных этой возрастной группы в основном встречается печеночная манифестация, церулоплазмин снижается в диагностически значимых пределах у 82 – 98% (36, 63, 98, 221).
В тоже время, в одном клиническом исследовании, где существенно пониженный уровень церулоплазмина отмечался лишь в 42,9% и еще в 28,6% немногим ниже нормы, авторы сделали вывод о малой информативности этого теста (151).
Аналогично уровню церулоплазмина в крови, общая концентрация меди в сыворотке при болезни Вильсона также обычно понижена (< 50 – 60 мг/дл; при норме 65 – 165 мг%)(118, 157). Содержание меди в сыворотке крови представляет собой сумму свободной, несвязанной и несвободной, связанной с церулоплазмином меди. При болезни Вильсона на фоне общего пониженного количества этого микроэлемента в крови доля свободной меди значительно повышается (> 3,9 мкмоль/л) по сравнению с контрольной группой у здоровых (норма: 0,8 – 1,9 мкмоль/л) и обычно существенно выше, чем 10 мг/дл. Определение концентрации не связанной с церулоплазмином меди в крови производится путем вычитания от суммарного параметра количества связанной меди. Однако в случае фульминантного гепатита при болезни Вильсона общее содержание меди отчетливо повышается (118).
В педиатрической практике одни авторы отмечают большую частоту отклонения этого параметра, что составляет от 75 до 96% случаев (36, 151). Некоторые этот тест даже выделяют как самый чувствительный (36). Но по наблюдениям других исследователей сниженный уровень меди в крови встречался только у 55% больных детей и они оценили диагностическое значение данного анализа как мало информативное (63).
Следующим методом лабораторной диагностики является определение количества меди в моче за 24 часа. При болезни Вильсона содержание меди в моче повышено > 100 мг/сутки, что встречается приблизительно у ¾ больных с симптомами заболевания. Выполнить этот тест несложно, поэтому он доступен, но требует тщательного соблюдения правил сбора материала, так как из-за этого не редко возникают ошибки из-за контаминации мочи.
Однако некоторые авторы подчеркивают, что у детей 24 часовой сбор мочи иногда показывает нормальное содержание меди. Это также может быть у женщин во время беременности, несмотря на активно протекающую болезнь, или когда женщина принимает гормональные контрацептивы. В случае серьезного подозрения на болезнь Вильсона, исследование целесообразно повторить на следующий день после двукратного приема с интервалом в 12 часов разовой дозы Д-пеницилламина (DPA). После приема DPA показательным в отношении подтверждения диагноза является увеличение количества меди в моче в разы.
Данные других исследований о чувствительности этого теста при диагностике болезни Вильсона у детей показывают, что диагностически значимое повышение содержание базальной меди в моче встречается в 73 – 100% случаев (36, 63, 98, 151). В некоторых исследованиях указаны средние значения и доверительные границы интервала полученных измерений: в серии больных из Германии колебания находились в пределах от 267,9 до 452,8 мг/сут. (36), среднее значение количества меди за сутки в моче у больных в канадском исследовании составило 11,5 мкмоль/дл. После приема DPA 9 из 16 больных имели повышение содержание меди не менее чем до 25 мкмоль/дл (т.е. более чем в 2 раза). В тоже время у остальных 7 (44%) детей концентрация меди не достигла доказательного уровня, хотя у всех них диагноз был подтвержден генетически (151).
Основным диагностическим тестом, окончательно подтверждающим диагноз болезни Вильсона, считается количественный анализ меди в ткани печени. В норме содержание этого микроэлемента в печени < 40 мг/г (15 – 30 мг/г) сухого веса печеночной ткани. В тоже время при болезни Вильсона обычно превышает 200 – 250мг/г сухого веса, достигая у некоторых больных 3000 мг/г. У гетерозиготных носителей генетического дефекта концентрация меди в печени занимает промежуточное место между здоровыми и больными, находясь в пределах между 50 и 250 мг/ г сухого веса. Имеются данные о содержании меди в печени отдельно у детей. Сообщается, что ее концентрация при болезни Вильсона может составлять от 250 до 1200 мкг/г (221). Вероятно в связи с меньшей продолжительностью заболевания по сравнению с взрослыми значения этого показателя у них ниже. Для определения концентрации меди используются методы атомной адсорбции и «mass»- спектрометрии (59, 118). Однако следует иметь в виду, что при некоторых заболеваниях печени, особенно сопровождающихся холестазом, в ткани печени тоже обнаруживается повышенное содержание меди, хотя при этом уровень в 250 мг меди на 1г сухого веса превышается незначительно.
С помощью биопсии печени можно получить возможность использовать и другие методы диагностики, а также определить стадию заболевания. При проведении исследования важно применять иглу из материала, не содержащего медь, а шприц ни в коем случае не заполнять солевым раствором. Это позволить исключить неправильные результаты исследования.
Гистологические данные на ранних стадиях болезни Вильсона соответствуют картине мелкокапельного или крупнокапельного ожирения, затем патологический процесс ведет к развитию фиброза и, наконец, цирроза печени.
Выявление болезни Вильсона на ранних стадиях заболевания вероятно до сих пор представляет трудную задачу для врачей общей практики, так как полученные данные гистологического исследования печеночных биоптатов у детей при первичной диагностике показывают, что большая часть уже имеет признаки тяжелого поражения печени вплоть до цирроза. Так в кубинской серии стеатоз без или с наличием фиброза, хронический гепатит и цирроз были обнаружены у 73% больных, и только у 27% – поражение печени с минимальными патологическими признаками или имелась нормальная структура ткани органа. Об этом свидетельствуют и другие данные: у 16 детей из Туниса цирроз был диагностирован в 8 случаях (18), в уже упомянутом выше клиническом исследовании из Турции из 33 детей по данным биопсии или вскрытия 18 больных имели цирроз печени, 9 хронический гепатит и 6 массивный некроз.
Для диагностики болезни Вильсона предпринимались попытки с помощью гистологических окрасок обнаружить избыток меди, но у детей по опыту некоторых авторов это удается редко (36). В частности гистологическая окраска родонином позволяет обнаружить родонин позитивные гранулы в гепатоцитах менее чем в 10% подтвержденных случаев болезни Вильсона (221). Другие авторы применяли окраску орсеином и в 88% случаев получили положительные результаты.
Специальное окрашивание при электронной микроскопии ткани печени позволяет выявить специфические изменения в митохондриях гепатоцитов. Однако считается, что их можно заметить только на ранних стадиях болезни, в период, когда при световой микроскопии обнаруживается жировая инфильтрация. В одном детском клиническом исследовании типичные признаки в митохондриях при электронной микроскопии находили в 93% случаев, но среди обследованных была большая доля бессимптомных больных (151). Поэтому оценить чувствительность этого диагностического теста у детей, особенно в продвинутых стадиях болезни представляется затруднительным.
Если имеются противопоказания к биопсии печени, клинический диагноз неясный, а биохимические тесты не показательны, то, возможно, провести также тест с изотопами меди. При этом радиоизотопном исследовании у здоровых лиц отмечается два пика радиоактивности в крови. У пациента с болезнью Вильсона второй подъем радиоактивности отсутствует. Тест служит также для дифференциальной диагностики между гомозиготными и гетерозиготными носителями генетического дефекта и для диагностики очень редкого варианта болезни Вильсона у больных с нормальным уровнем церулоплазмина. Правда, опытом такого рода исследований располагают только некоторые центры, а сама методика очень дорогая (118).
При фульминантном гепатите вследствие болезни Вильсона, когда время для диагностики очень ограничено, при клинической картине острой печеночной недостаточности неясной этиологии, на данное заболевания указывают гемолитическая анемия с отрицательной пробой Кумбса, значительно повышенный уровень выделения меди с мочой и заметное снижением активности щелочной фосфатазы в крови.
Оценку уровня щелочной фосфатазы проводят соответственно возрастной норме этого показателя. Необычно «нормальная» активность этого фермента при фульминантном гепатите в результате болезни Вильсона дало основание некоторым гепатологам использовать коэффициент отношения билирубина к активности щелочной фосфатазы как дополнительный диагностический тест. Отношение < 2 расценивается как весьма вероятное относительно подтверждения диагноза фульминантной формы болезни Вильсона, хотя соотношение > 2 также не исключает этого диагноза (118).
Молекулярно-биологический метод диагностики. Сегодня имеется возможность надежно установить диагноз болезни Вильсона с помощью молекулярного мутационного анализа. Для этого используют ДНК лейкоцитов. Так как сегодня сообщается о более 260 мутаций в гене болезни Вильсона и большая часть из них пока еще не признаны, как имеющие отношение к нарушению метаболизма меди в организме, то требуется осторожность и определенный опыт при интерпретации полученных данных. Чтобы получить надежный результат, раньше было важным подобрать оптимальный праймер для ПЦР (соответствующий самой распространенной мутации в данной популяции). Если с выбором праймера удавалось «угадать», то диагноз можно было подтвердить быстро и относительно недорого. В настоящее время предлагается алгоритм мутационного анализа, значительно упрощающего исследование и уменьшающего затраты на его проведение (33).
Лечение. При болезни Вильсона речь идет заболевании, которое без лечения, неизбежно ведет к смерти. В тоже время сегодня медицина располагает высоко эффективными методами терапии, которые полностью устраняют влияние болезни на ожидаемую продолжительность жизни больного.
Однако современные методы лечения позволяют только связывать избыток меди и вывести его из организма, для того чтобы предотвратить необратимые повреждения внутренних органов. Так как в основе болезни лежит генетический дефект, то лечение пожизненное и закончить его можно только тогда, когда больному пересаживают печень.
Лечение больного начинается сразу же после установления диагноза. Оно показано как больным с уже развившимися симптомами болезни, так и тем, у кого симптомов еще нет. Основным препаратом, в использовании которого накоплен самый большой практический опыт, является Д-пеницилламин (DPA, торговые названия Metalcaptase, Купренил). Это комплексобразующее соединение, способное образовывать хелатные соединения с ионами многих металлов, в том числе с медью.
Многолетние наблюдения позволили определить стандартную схему назначения DPA в суточной дозе 20 мг/кг (но не более 2 г), которая делится на 3 приема. Таблетки принимают до еды. Однако существует и другая точка зрения, когда в первые 2 – 3 месяца рекомендуются очень высокие дозы (приблизительно 30 мг/кг, но не более 3 г). Напротив, третьи предлагают начинать с половинной дозы (приблизительно 10 мг/кг) из-за боязни развития побочных явлений, в первую очередь, аллергических реакций. С учетом последнего обстоятельства дополнительно можно назначить антигистаминные препараты.
Так как DPA является антагонистом пиридоксина, одновременно больной должен принимать витамин В6.
До начала лечения и в его процессе за больным устанавливается наблюдение с периодическим углубленным обследованием в рамках выработанного протокола. Перечень основных клинических, лабораторных и инструментальных исследований представлен в таблице 25.
Таблица 25.
Лабораторно-инструментальные методы для диагностики и мониторинга за болезнью Вильсона.
Метод обследования
При первичной диагностике
Во время лечения
Офтальмологическое исследование в щелевой лампе (Кольцо Кайзера-Флейшера)
+
+
Неврологический статус
+
+
Клинический анализ крови
+
+
Проба Кумбса
+
–
Биохимические тесты: АлАТ, АсАТ, билирубин, общий белок, протеинограмма, протромбин, креатинин, мочевина
+
+
Биохимические тесты обмена меди: церулоплазмин, содержание меди в сыворотке крови (общая, связанная с церулоплазмином и свободная фракции меди), содержание меди в моче за 24 часа
+
+
Общий анализ мочи
+
+
Радиоизотопное исследования обмена меди
+
–
Биопсия печени с последующим гистологическим (в т.ч. окраска орсеином или родонином), гистохимическим и электронно-микроскопическим исследованием
Эффективность лечения лучше всего контролировать по ее содержанию в ткани печени, но для этого необходимы повторные биопсии, не всем доступен метод определения концентрации меди в печеночной ткани. Среди так называемых неинвазивных тестов наиболее точно позволяет оценить успехи в нормализации обмена меди в организме это определение в крови свободной меди, концентрацию которой рекомендуется поддерживать во время лечения постоянно ниже 10-20 мг/дл. Вторым важным параметром является мониторинг за выделением меди с мочой, оптимальным снижением которой считается уровень приблизительно до 0,5 мг в сутки. Если после однодневной паузы в приеме DPA выделение меди с мочой за 24 часа меньше 100 мг, можно считать выбранную дозу препарата правильной (118).
В результате эффективного лечения DPA вначале наступает нормализация тестов, характеризующих обмен меди, затем уменьшается выраженность отдельных клинических симптомов или их полное исчезновение (см. рис. 1 и 2). Однако положительная динамика клинических проявлений болезни Вильсона в процессе терапии отмечается только через несколько месяцев, – иногда лет. Более того, приблизительно у 30% больных вначале лечения наблюдается ухудшение или даже появляются неврологические симптомы, которых до этого у больного не было. Это не следует рассматривать как плохой прогноз или как неадекватная выбранная схема терапии (118).
Улучшение клинико-лабораторных данных является основанием для перехода на поддерживающее лечение DPA, приблизительно в половинной дозе от первоначальной.
При пожизненной терапии DPA вопрос о побочных действиях этого препарата и своевременном их распознавании очень важен. Поэтому лечение минимально возможной, но в тоже время эффективной дозой DPA является очень актуальным.
Приблизительно у ⅓ больных в первые 5 недель от начала лечения развивается аллергия (118, 171). В таких случаях рекомендуют уменьшить дозу DPA и продолжить терапию, а затем очень медленно ее увеличивают, принимая вместе с антигистаминными препаратами. Также дополнительно на 2 недели можно назначить кортикостероиды в средней дозировке, после чего дозу гормонов постепенно уменьшают до полной их отмены (118).
Кроме реакции гиперчувствительности, описаны другие побочные действия DPA, но встречаются они редко. Среди них эозинофилия, тромбоцитопения, лейкопения, анемия (апластическая и гемолотическая), агранулоцитоз; нефрит, гепатит, внутрипеченочный холестаз, панкреатит; полимиозит, дерматомиозит, миостения. Побочные действия при лечении могут вести даже к смерти. Однако, в общем и целом только 2% больных не переносят DPA. В случаях полной интолерантности назначаются другие препараты.
Ни в коем случае лечение нельзя прерывать. Оно, как уже отмечалось выше, должно быть пожизненно постоянным. Необоснованная отмена терапии врачом, например, на основании нормализации функциональных печеночных проб и показателей обмена меди, или самовольный отказ больного от лечения ведут к развитию фульминантного гепатита с крайне неблагоприятным прогнозом для жизни. Обычно через несколько месяцев после его прекращения наблюдается бессимптомное повышение трансаминаз, но вскоре внезапно, без видимой причины у больного возникает молниеносно прогрессирующая острая печеночная недостаточность.
Болезнь Вильсона не является противопоказанием для беременности. Более того, в этот период обмен меди в организме стабилизируется. По этой причине ранее рекомендовалось в первый триместр беременности лечение DPA прервать. В тоже время, накопленные за последние годы данные показали, что DPA не обладают тератогенным действием, поэтому эти рекомендации изменены. Во время беременности DPA необходимо продолжать принимать, только, как советуют некоторые авторы, в этом случае дозу препарата целесообразно снизить до 750 мг/сут (118). Однако лактация является противопоказанием для приема DPA, а так как терапию остановить нельзя, ребенка приходиться переводить на искусственное вскармливание.
Среди альтернативных DPA препаратов имеется еще один с аналогичным механизмом действия – тетраэтилентетрамин [триентин] (tetraethylene tetramine dihydrochloride, trientine, EDTA), который тоже способен образовывать хелатные комплексы с ионами меди. Переносимость тетраэтилентетрамина хорошая. Его назначают в суточной дозе от 1200 до 2400 мг, которую делят на 3 приема. Препарат также необходимо употребить приблизительно за 1 час до приема пищи. Важнейшими побочными действиями триентина являются гипохромная анемия и протеинурия.
Возможно в будущем в этой группе наравне с уже упомянутыми будут аmmonium tetrathiomolybdate и dimercaptopropanol. Первый из них показал высокую эффективность, но число пролеченных им больных очень маленькое. Поэтому можно считать, что испытания еще не завершены. Dimercaptoproranol пока назначают как вспомогательный препарат при лечении DPA, когда неврологические симптомы или психические проявления у больного сохраняются без динамики во время монотерапии.
Вторая группа препаратов – это производные цинка. Цинк блокирует всасывания меди из кишечника за счет индукции в слизистой metallothionein (21). В результате резорбция меди уменьшается, образовавшиеся из микроэлементов комплексы выделяются со стулом.
Некоторые авторы рассматривают эти две группы препаратов с различным механизмом действия как альтернативные (118). Другие считают, что хелатобразующие субстанции являются облигатными средствами или, по крайне мере, препаратами первого ряда. Тогда как препараты цинка следует использовать только в дополнение к первой группе, позволяющие максимально снизить дозу DPA и таким образом избежать побочных действий. Другая ситуация для назначения цинка – замена препаратов первого ряда, после клинического улучшения и нормализации параметров обмена меди (171).
Рекомендуемая доза цинк ацетата или цинк сульфата составляет от 45 до 150 мг в сутки. Ее также делят на 3 приема, каждый раз прием препарата должен быть по крайне мере за час до еды. По мнению некоторых авторов их целесообразно применять, прежде всего, у детей. Особенно хорошо без побочных явлений переносится цинк ацетат. Основное побочное действие препаратов цинка боли в животе, что обычно удается избежать, если первый прием переносится на время между завтраком и обедом (118).
Однако следует считать, что оптимальная схема лечения болезни Вильсона у детей еще не определена. Несмотря на привлекательность применение препаратов цинка у детей из-за отсутствие серьезных побочных действий, на практике чаще используется DPA (18, 98, 118).
Рекомендовать диетическое питание при болезни Вильсона сложно, так как существенно ограничить поступление меди с продуктами почти невозможно. Печень и улитки особенно богаты этим микроэлементом. В тоже время при хорошо отрегулированном обмене меди при болезни Вильсона в результате лечения в небольших количествах разрешается употреблять любые продукты питания.
При фульминантном гепатите при болезни Вильсона прогноз абсолютно неблагоприятный и больные умирают приблизительно в течение 2 месяцев. Единственная возможность спасти больного это сделать ему пересадку печени, но перед операцией проводится подготовка, представляющая собой интенсивную терапию как при фульминантном гепатите любой другой этиологии. Острая печеночная недостаточность в результате массивного некроза печени является единственным показанием для трансплантации этого органа. Трансплантация печени совершенно бесполезна, если доминирует неврологическая симптоматика, угрожающая жизни больного, а признаков печеночной недостаточности нет. В результате успешной пересадки печени больной полностью излечивается от болезни Вильсона (118).
VII. Гемохроматоз.
Сегодня термином гемохроматоз обозначают заболевание, в результате которого в организме человека прогрессивно увеличивается запас железа. Хроническая перегрузка железом ведет к его генерализованному отложению в различных тканях. Прежде всего, железо в избытке накапливается в паренхиматозных клетках печени и сердца, в секреторных клетках, половых железах и некоторых других органах, вызывая в дальнейшем их повреждение. Наследственный гемохроматоз обусловлен аутосомным рецессивным геном, сцепленным с локусом главного комплекса гистосовместимости в коротком плече хромосомы 6, т.е. проявляется заболевание только у тех лиц, которые приобретают две копии мутированного гена, по одному от каждого из родителей.
Другие заболевания также могут вести к гемохроматозу. По мнению некоторых авторов, эти случаи рассматривают как вторичный гемохроматоз (136). Другие считают, что гемохроматоз при хронической гипохромной гемолитической анемии, сцепленной с Y-хромосомой, талассемии и атрансферринемии также является первичным.
Очень редко гемохроматоз может возникнуть из-за продолжительного парэнтерального или энтерального приема железа, а также при передозировке витамина С. Вторичный гемохроматоз наблюдается у африканцев банту в результате употребления ими местных алкогольных напитков и эфиопов из-за использования в качестве пищи тэффа (разновидность просо).
Генетика и патогенез наследственного гемохроматоза. Ген, мутация в котором ведет к гемохроматозу, недавно был обнаружен в главном комплексе гистосовместимости (HLA) и в настоящее время обозначается HFE геном. Продуктом этого гена является 343 фрагмент трансмембранного гликопротеина I типа, который гомологичен антигенам MHC I класса и связан с b2-микроглобулином (56). Гемохроматоз проявляется только у тех лиц, которые приобретают две копии мутированного гена, по одному от каждого из родителей.
Среди людей североевропейского происхождения около 65 – 95% больных с клинически выраженным гемохроматозом имеют точечную мутацию HFE гена в области перехода последовательности нуклеотидов от гуанина к аденину. Это ведет к замене в позиции 282 цистеина на тирозин [C282Y-мутация или Tyr282Cys] в структуре кодируемого им полипептида (56, 127). Значение второй мутации в позиции 63 HFE гена с заменой гистидина на аспартат (H63D-мутация) для возникновения наследственного гемохроматоза до сих пор до конца неясное. Эту мутацию обнаруживают приблизительно у 5% лиц в общей популяции. Гомозиготные носители этой мутации не относятся к группе риска в отношении появления у них наследственного гемохроматоза. Однако, у тех лиц, у которых выявляется гетерозиготный состав обоих мутаций (C282Y и H63D), также относятся к группе относительно высокого риска развития гемохроматоза (≈ 4% случаев) (56, 118). Статус гетерозиготного носителя может также играть негативную роль при одновременно имеющемся другом тяжелом заболевании печени, например, при гепатите С. При этом обмен железа в организме существенно ухудшается и в печени отмечается избыток этого микроэлемента, хотя в значительно меньшей степени, чем при манифестном гемохроматозе. В свою очередь это негативно влияет на эффективность лечения вирусного гепатита С интерфероном.
Гемохроматоз является одним из наиболее частых наследственных заболеваний в Европе. В европейской популяции 10% лиц являются гетерозиготными в отношении гена, из-за которого возникает заболевание (157), в США – 6,7%. Последние исследования также показали, что частота гомозиготных вариантов мутаций в отношении гемохроматоза у лиц североевропейского происхождения находится в пределах от 0,3 до 1% (136, 157). Мутация в аллели Tyr282Cys среди ирландцев и басков еще выше - у 10 и 30,4% соответственно. В тоже время среди больных гемохроматозом в южной Европе подобную мутацию имеют менее 65% обследованных, а в Африке, Азии и Австралии ее пока вообще не обнаружили (127).
Недавно было описано взаимодействие HFE гена с рецептором трансферрина. В отличие от природного продукта, кодируемого нормальным геном, мутант HFE [Tyr282Cys] не прочно связывается с рецептором трансферрина и не снижает сродство трансферрина к железу (57, 144). Таким образом, в присутствии мутированного Tyr282Cys продукта поступление железа внутрь клеток облегчается. Было также установлено, что при базовой pH растворимый рецептор трансферрина и продукт HFE гена на поверхности клетки компактно связаны. При изменении pH в кислую сторону, например в эндосомах или других внутриклеточных вакуолях они разъединяются (105). Вероятно, так и происходит в организме человека. Продукты HFE гена попадают в клетку вместе с комплексом трансферрин-трансферрин, а затем этот белок диссоциирует из этого комплекса при кислых значения рН во внутриклеточных вакуолях. Последние данные также показали, что комплекс HFE белка с рецептором трансферрина возникает посредством одиночной связи, таким образом, образуется димер. Образование димера вызывает асимметрию рецептора трансферрина в сторону наиболее оптимального участка связывания этого белка с железом. Именно поэтому уменьшается сродство трансферрина с железом (105). В противоположность мутации Tyr282Cys, замена His63Asp не препятствует связи с b2-микроглобулином или экспрессии на оболочке клетки (210).
Нарушения функционирования между рецептором трансферрина и продуктом мутированного HFE гена может содействовать поступлению повышенного количества железа и его избыточному накоплению в клетках некоторых тканей и органов. Но каким образом эта мутация ведет к повышению абсорбции железа из кишечника остается неясным, так как первичный дефект имеется не в печени, в слизистой гастроинтестинального тракта.
Иммуногистохимический анализ показал, что продукты мутированного HFE гена обнаруживаются в определенных участках тонкого кишечника, преимущественно в 12 перстной кишке (145). Однако клетки слизистой этого отдела кишечника, характерным признаком которых являются наличие у них глубоких крипт, не участвуют в регуляции абсорбции железа. Таким образом, точный механизм, через который HFE мутация ведет к повышенной абсорбции железа из кишечника, требует разъяснений.
Цепь патофизиологических эффектов при гомозиготном гемохроматозе оборачивается перегрузкой организма железом. Этот микроэлемент регулирует функции многих клеточных ферментов. При избытке железа происходит нарушение в работе некоторых энзимов, участвующих в окислительно-восстановительных реакциях, что вызывает повышенное образование свободных кислородных радикалов, которые являются очень токсичными.
При клинически выраженном заболевании содержание железа в организме может повышаться до 20 – 40 г в сравнении с 1 – 4 г в норме. Эта перегрузка организма железом вызвана чрезмерным поступлением железа из желудочно-кишечного тракта приблизительно от 2 до 5 г в день в сравнении с 1 мг у здорового человека. Развернутая картина болезни наблюдается только у гомозиготных лиц. Однако некоторые гетерозиготные лица также могут иметь небольшие изменения лабораторных тестов в отношении метаболизма железа в организме. У тех, кто имеет гомозиготный дефект, внешние проявления заболевания значительно варьируют, что, по всей видимости, обусловлено факторами внешней среды и характером питания.
Также остаются неясными и последующие фазы патологического процесса, после того как железо в избыточном количестве поступает в организм человека. В частности точно не определены все внутренние органы и ткани, которые повреждаются в результате перегрузки железом. Нарушений в функционировании рецепторов ферритина и трансферрина, ответственных за поступление железа в определенные типы клеток, недостаточно, что объяснить патогенез этой болезни. В этой связи определенный интерес представляет тот факт, что у больных с гемохроматозом имеется значительное увеличение в плазме фракции железа, несвязанной с трансферрином. Концентрация этой фракции постепенно снижается во время лечения и возможно этой составляющей также может быть обусловлено поражение различных органов и тканей (78).
Диагноз. Многие десятилетия гемохроматоз диагностировали обычно у больных с симптомами болезни. Однако будущие усилия должны быть сконцентрированы на ранней диагностике, потому что своевременно начатое лечение очень эффективно и, прежде всего, у тех, у кого еще не развились поздние органные осложнения.
Ранняя диагностика наследственного гемохроматоза у лиц, которые еще не имеют симптомов цирроза и других органных осложнений в результате большой и длительной перегрузки железом организма, может быть осуществлена или с помощью биопсии, или при проведении генетического анализа. Такой подход позволяет более чем в 4 раза чаще поставить диагноз еще на бессимптомной стадии развития этой болезни (137). Существенно улучшает диагностические возможности применение на практике определения ферритина в крови как неинвазивного и высоко чувствительного теста, позволяющего оценить уровень накопления железа. Важно отметить, что большинство больных без симптомов выявляются при скрининговом обследовании членов семьи больного с уже диагностированным гемохроматозом. Последних, таким образом, относят к важнейшей целевой группе при планировании массовых обследований.
Является ли биопсия печени при наличии новых генетических маркеров излишней остается спорным. Пока большинство специалистов используют данные биопсии печени как золотой стандарт для диагностики гемохроматоза. Но в ряде случаев это исследование не требуется, например, у больных с выраженным циррозом печени, при котором биопсия печени не даст дополнительной информации. Также у тех, кто имеет незначительное повышение насыщенности трансферрина и ферритина в крови, и при этом у них отсутствуют какие-либо клинические проявления поражения печени. Однако следует иметь в виду, что только биопсия печени показывает истинную выраженность патологического процесса в печени из-за перегрузки органа железом, выраженность фиброза и наличие сопутствующих заболеваний печени. Поэтому показания к биопсии индивидуальны каждому больному.
Определение насыщения трансферрина железом в крови, а также ферритина являются в настоящее время более простыми и доступными лабораторными тестами для ранней диагностики. Оба указывают на заболевание, но только у лиц с определенным «стажем» болезни, когда уже имеется накопление железа такой степени, что значения этих лабораторных показателей начинают превышать норму. Убедительно доказано, что насыщенность трансферрина Fe является более чувствительным маркером для диагностики гемохроматоза. Ферритин повышается только тогда, когда имеется очень большая перегрузка железом организма (47). Определение генетических маркеров позволяет надежно подтвердить диагноз.
Большинство авторов сегодня также подсчитывают индекс железа печени (114). Некоторые практические рекомендации об использовании лабораторных тестов и их диагностическое значение отражены в работе Rasenack J., 2003 (157), хотя автор в качестве первого шага предлагает использовать наряду с определением содержания железа в крови также концентрацию содержащего железа белка – ферритина, а не насыщения железом трансферрина. Только если значение этих тестов оказываются повышенными, то определяется насыщенность трансферрина железом в сыворотке крови. Окончательно диагноз гемохроматоза автором предлагается подтвердить исследованием концентрации железа в ткани печени с подсчетом печеночного индекса (Fe мг в 1г сухого веса печени / возраст ´ 56).
В таблице 26 отражены нормальные значения основных лабораторных тестов, которые используются в диагностике гемохроматоза, и их изменения, характерные при генетических аномалиях, связанных с гемохроматозом, и алкогольной болезни.
Наконец наследственный гемохроматоз можно подтвердить также при генетическом анализе, когда обнаруживают одну из мутаций. Если мутация С282G гомозиготная, то диагноз гемохроматоза бесспорен. Однако гемохроматоз не исключается, если эта мутация отсутствует. Число таких случаев находится в пределах от 10 до 35%, среди европейских стран особенно часто это встречается в Италии.
В тоже время при поздней кожной порфирии, которая до сих пор также лечится только кровопусканиями, мутация встречается С282G очень часто.
Для постановки генетического теста применяют метод ПЦР.
Таблица 26.
Характеристика лабораторных показателей при наследственном гемохроматозе (157).