Кислород, в зависимости от формы его нахождения в галогенидах, может быть определен различными методами.
Фторидный метод.
Основан на способности фтора замещать кислород в окислах металлов. Реакция фторирования протекает достаточно быстро (20-30 мин.) при температурах 300-4000С. В качестве реагентов, помимо газообразного фтора, могут применять вещества, содержащие высокоактивный фтор, такие как KBrF4 или CsBrF6. Замещение кислорода сопровождается его выделением в молекулярной форме:
2CaO+2СsBrF6→ 2CaF2 +O2↑+2CsBrF4
Объем выделившегося газа измеряют при атмосферном или пониженном давлении. С помощью этого метода возможно определение кислорода в любых галогенидах щелочных и щелочноземельных металлов, если при взаимодействии реактива с веществом не образуются летуче кислородные соединения, например оксигалогениды. Чувствительность определения зависит от чистоты реактива и количества кислорода, находящегося на стенках применяемой аппаратуры, и составляет 0,1-0,003 вес.%.
Метод изотопного разбавления.
При анализе данным методом не требуется полноты экстракции кислорода из пробы. Сущность метода состоит в следующем. Анализируемое вещество плавится в тигле вместе с образцом, содержащим известное количество кислорода, изотопный состав которого отличается от природного. Предполагается, что в течение некоторого промежутка времени изотопная концентрация кислорода во всем объеме печи выравнивается. Затем следует отбор газа и определение изотопного состава кислорода. Содержание кислорода в анализируемом образце рассчитывают по формуле:
Cx=
где Gd-количество кислорода, содержащегося в дозировочном образце
Чувствительность метода ограничивается из-за флуктуации величины поправки холостого опыта.
Физические свойства большинства галогенидов (температура плавления, летучесть, возможность восстановления окислов углеродом при температуре меньше 2500К) позволяют достаточно широко использовать метод изотопного разбавления. Однако наиболее целесообразно применение этого метода для анализа фторидов, в которых определение кислорода радиоактивационными методами затруднено вследствие присутствия фтора.
Метод радиоактивационного определения.
Для данного метода мог у т быть использованы многие ядерные реакции. Наибольшее применение получили следующие: на быстрых нейтронах -
016(n,р)N16; N16Т 1/2 - 7,35 сек;
на γ-квантах -
016(γ,n)015; 015 T 1/2 - 2,05 сек;
на заряженных частицах -
016(α, pn)F18;
O16(He3,p)P18;
O16(t,n)F18;
O16(p,n)F18;F18T 1/2 - 112,2 мин.
Щелочные, щелочноземельные металлы, хлор и йод не образуют при облучении изотопов, которые бы мешали определению кислорода. Бром при γ-активации образует изотоп с периодом полураспада, близким к 2 мин. Конечные продукты ядерных реакций кислорода и фтора с быстрыми нейтронами, заряженными частицами и γ-квантами идентичны. По этой причине раздельное определение кислорода и фтора таким методом невозможно. Исключение составляет определение кислорода во фториде лития с помощью облучения реакторными нейтронами по сопряженной реакции
Li6(n,α)t; Оl6(t,n)F18.
Ввиду малого сечения реакции F19(n,2n)F18 можно определять кислород во фториде лития с чувствительностью до 10-3%. Как правило, радиохимическое выделение не представляет самостоятельной задачи при проведении анализов радиоактивационными методами.
Метод активации быстрыми нейтронами, обладающий сравнительно невысокой чувствительностью10-3%, имеет преимущество перед другими радиоактивационными методами, заключающееся в том, что наведенную активность удается регистрировать прямыми инструментальными методами без радиохимического выделения изотопа. Чувствительность методов активации заряженными частицами составляет10-4-10-7%; γ-квантами 10-2-I0-4%.[1]
Масс-спектрометрия.
Применение масс-спектрального окончания приопределении ГП позволяет значительно снизить пределы обнаружения и повысить точность анализа. Благодаря разработке методики очистки поверхности образца перед анализом непосредственно в источнике ионов времяпролетного тандемного лазерного масс-рефлектронадостигнуты рекордные пределы обнаружения ГП для масс-спектральных методов анализа. Масс-спектральный метод пока является достаточно длительным, трудоемким и дорогим методом анализа.
ИК-спектроскопия.
Очень высокой чувствительностью обладают методы ИК-спектроскопии, применяемые при определении кислорода в кремнии и полупроводниковых материалах. Однако метод связан с использованием сложного оборудования, применим только для анализа твердых веществ, прозрачных в инфракрасном диапазоне излучения, и не всегда универсален из-за широкого набора спектров поглощения и отсутствия сведений о коэффициентах экстинкции.[2]
К методам определения общего содержания кислорода относятся высокотемпературное бромированиеKBrF4 и восстановительное плавление в присутствии углерода.
Достоинства восстановительного плавления: он проще, обладает большейвоспроизводимостью и позволяет использовать промышленную аппаратуру.
Метод основан на переводе кислорода образца в газовую фазу в форме окиси углерода при плавлении в вакууме или в среде инертного газа в графитовом тигле. Температура процесса восстановления может быть приблизительно определена на основании термодинамических расчетов.[1]
Для методов, используемых на практике для определения общего содержания кислорода, характерно следующее:
- среда в реакционной камере - вакуум или поток инертного газа (азот, аргон, гелий);
- графитовый тигель (одноразового применения) используется только для одной испытываемой навески или накопительный тигель (тигель многоразового использования) - для нескольких последовательно испытываемых навесок;
- реакционная среда - сухая (без жидкой фазы), т.е. в графитовый тигель помещают одну испытываемую навеску и восстановление проводят в твердофазном состоянии, т.к. анализируемый металл не плавится.
С целью ускорения восстановления некоторых металлов используют металлическую ванну (восстановление в жидкофазном состоянии). Для этого рекомендуется сначала приготовить ванну из плавкого металла (например платины, олова, железа, никеля), способного растворять как углерод, так и металл в испытываемой навеске. Нагрев непрерывный, если испытываемую навеску помещают в тигель, предварительно нагретый до температуры реакции; при этом восстановление происходит в несколько минут.
Нагрев импульсный, если холодный тигель с испытываемой навеской с помощью мощного импульса энергии нагревают за несколько секунд до пиковой температуры 3000 °С, при которой восстановление происходит очень быстро.
Определение кислорода по измерению содержания СО и СО2 осуществляют различными методами. Во всех методах используют устройства для химической конверсии, обеспечивающие полное превращение оксидов углерода в СО или СО2. Обычно используют следующие аналитические методы: объемный (для монооксида углерода); хроматографии (для монооксида углерода); инфракрасного поглощения (для диоксида и монооксида углерода); теплопроводности (для монооксида и диоксида углерода); кулонометрии (для диоксида углерода).
При непрерывном нагреве и восстановлении в твердом состоянии реакция восстановления может протекать медленно, и время полного восстановления оксидов зависит от содержания кислорода. Поэтому невозможно определить условия определения кислорода для каждого из анализируемых металлов, сплавов и карбидов и каждого типа применяемых приборов (установок).
Рекомендуется путем выполнения предварительных испытаний определять оптимальные условия анализа для конкретного типа материала и предполагаемого в нем содержания кислорода. С этой целью на одной и той же пробе проводят ряд последовательных испытаний, ускоряя процесс ее восстановления увеличением температуры и (или) времени реакции вплоть до момента, когда содержание измеряемого кислорода достигнет максимального и постоянного значения. При восстановлении можно также изменять другие параметры (например, металл ванны).
Для обеспечения установленных рабочих условий рекомендуется применять только, имеющие сертификаты, эталонные образцы (стандартные образцы по нормативным документам) того же типа, что и материал пробы. [3]
Особенности галогенидов щелочных и щелочноземельных металлов, как объектов анализа методом восстановительного плавления:
1. Упругость паров этих солей при температуре восстановления окислов соответствующих металлов велика, и потому их восстановительное плавление можно проводить только в атмосфере инертного газа.
2. Пары галогенидов весьма агрессивны и вызывают коррозию аппаратуры. Поэтому кварцевые детали печи следует охлаждать проточной водой.
3. В литературе нет сведений о поглощении выделенной из образца окиси углерода возгонами галогенидов или их парами. Испарение галогенидов (но не окислов металлов) практически не сказывается на результатах анализа, если оно не вызывает механического уноса кислородсодержащих примесей.
Применение этого метода ограничивается галогенидами, обладающими малой летучестью при температуре экстракции газов (1900-2000 К), такими, как фториды щелочноземельных металлов, хлориды кальция, стронция и бария, а также бромиды кальция и бария. [1]
При протекании реакции между различными оксидами с углеродом может образовываться не только СО, но и другие летучие соединения, которые затрудняют анализ.
a) 1/m MenOm + C→n/m Me + (CO)
b) 1/(m-n) MenOm+ C→n/(m-n) (MeO) + (CO)
c) Bi2O3, Cu2O, PbO, NiO,WO2, FeO, MoO2
MenOm + (CO)→n/m Me + (CO2)
d) Au2O3, Ag2O, или оксиды платиновой группы
2/m MenOm→2n/m Me + (O2)
e) Sb2O3, PbO, In2O3
MenOm→(MenOm).
Также существует проблема сосуществования СО и СО2 при определенных условиях. Это следует учитывать при определении точности в определении кислорода.[4]
Также на результаты анализа влияют сами графитовые тигли. Выделение кислорода в пористых тиглях протекает гладко с отличной повторяемостью, а в плотных тиглях кинетика реакции становится неустойчивой, происходит ухудшение пиков. Чтобы получить лучшие результаты очень важно, чтобы внутренние тигли были с пористой губчатой морфологией, которая предотвращает накопление газообразных продуктов реакции на поверхности раздела расплав / графита.[5]
Механизм восстановления можно представить состоящим из следующих стадий:
1) насыщение расплава углеродом: С(гр)→[C]
2) адсорбция атомов углерода на межфазной поверхности включение — расплав: [С]→Садс;
4) десорбция атомов R с поверхности включений в расплав:
5) образование пузырьков СО на оксидных частицах в расплаве:
СОадс→Сгаз;
6) флотация связки пузырек - включение на поверхность расплава, отрыв пузырьков от включений и переход в газовую фазу;
7) перенос газообразных продуктов реакции потоком гелия в газодинамической системе анализатора.[6]
Вывод
В результате можно сделать вывод о том, что на правильность будут влиять следующие параметры:
- образование летучих соединений и CO2
- масса навески
- изменение состава ванны
- способ упаковки
Факторный эксперимент.
3.1. Fe2O3
Параметр оптимизации: случайная погрешность
Факторы: температура, масса образца, ванна.
Основной уровень: Т=4kW; m=40 мг; ванна – никель-олово
Интервалы варьирования:
- для температуры ±2kW
- для массы ±20мг
- для ванны –Ni, +Ni-Sn-Sn.
Таблица №9
Рабочая матрица планирования
№ опыта
Порядок реализации
Х1 (Т, kW)
X2 m (мг)
Х3 ванна
Ni
Ni
Ni
Ni
Ni-Sn-Sn
Ni-Sn-Sn
Ni-Sn-Sn
Ni-Sn-Sn
9 (основной уровень)
Ni-Sn
Таблица № 10
Рабочая матрица планирования в кодированных значениях.
№ опыта
Порядок реализации
Х1 (Т, kW)
X2 m (мг)
Х3 ванна
-
-
-
+
-
-
-
+
-
+
+
-
-
-
+
+
-
+
-
+
+
+
+
+
9 (основной уровень)
Ni-Sn
Проведение эксперимента
Таблица №11
Результаты
№ опыта
№ реализации
X0
Х1
Х2
X3
x1x2
x1x3
x2x3
x1x2x3
Y1
Т
m
ванна
+
-
-
-
+
+
+
-
0,805
+
+
-
-
-
-
+
+
0,569
+
-
+
-
-
+
-
+
0,739
+
+
+
-
+
-
-
-
0,532
+
-
-
+
+
-
-
+
0,554
+
+
-
+
-
+
-
-
0,716
+
-
+
+
-
-
+
-
0,659
+
+
+
+
+
+
+
+
0,671
0,789
Обработка результатов
Вычисляем коэффициенты регрессии.
b0 =
bi=
bij =
bijl =
и.т.д
Таблица № 12
Коэффициенты регрессии
b0
b1
b2
b3
b12
b13
b23
b123
0,656
-0,034
-0,0056
-0,0056
-0,015
0,077
0,020
-0,022
Проверяем значимость коэффициентов
Вычисляем дисперсию параметра оптимизации S2y
S2y=
Проверяем гипотезу о значимости коэффициентов регрессии по t – критерию Стьюдента. Для этого рассчитываем доверительный интервал для коэффициентов регрессии Δb
Δb=±t*Sb
Sb =
Таблица №13
Проверка значимости коэффициентов
основной уровень
Y1
Y ср
m-1
S^2
Sb^2
Sb
Δb
0,789709
0,74134
0,00108
0,00013
0,011626
0,032321
0,723662
0,726395
0,707733
0,759247
По результатам проверки получаем, что значимыми являются коэффициенты: b0,b1, b13.
Т.е. на параметр оптимизации будет влиять температура и температура-ванна. Коэффициентb1 отрицательный, поэтому чем меньше температура тем лучше.
Получаем уравнение:
y=0.656-0.034x1+0.77x1x3 (в кодированных значениях)
y=2.264-0.017x1+0.385x1x3-1.54x3 (в не кодированных)
Проверяем адекватность линейной модели.
Формальными признаками, по которым можно установить неадекватность линейной модели являются:
1. Незначимость хотя бы одного из эффектов взаимодействий
2. Незначимость суммы коэффициентов при квадратичных членах.
Оценкой этой суммы служит разность между b0 и значением параметра оптимизации в центре плана Ȳ0. Если эта разность превосходит ошибку опыта SȲ = , то линейная модель неадекватно описывает исследуемый процесс.
Ȳ-b0
SȲ
0,085583
0,032884
Линейная модель неадекватно описывает исследуемый процесс.
3.2. Al2O3
Параметр оптимизации: случайная погрешность
Факторы: температура, масса образца, ванна.
Основной уровень: Т=5.5kW; m=50 мг; ванна – никель-олово
Интервалы варьирования:
- для температуры ±0.5kW
- для массы ±10мг
- для ванны –Ni, +Ni-Sn-Sn.
Таблица №14
Рабочая матрица планирования
№ опыта
Порядок реализации
Х1 (Т, kW)
X2 m (мг)
Х3 ванна
Ni
Ni
Ni
Ni
Ni-Sn-Sn
Ni-Sn-Sn
Ni-Sn-Sn
Ni-Sn-Sn
9 (основной уровень)
5.5
Ni-Sn
Таблица № 15
Рабочая матрица планирования в кодированных значениях.
№ опыта
Порядок реализации
Х1 (Т, kW)
X2 m (мг)
Х3 ванна
-
-
-
+
-
-
-
+
-
+
+
-
-
-
+
+
-
+
-
+
+
+
+
+
9 (основной уровень)
5.5
Ni-Sn
Проведение эксперимента
Таблица №16
Результаты
№ опыта
№ реализации
X0
Х1
Х2
X3
x1x2
x1x3
x2x3
x1x2x3
Y1
Т
m
ванна
+
-
-
-
+
+
+
-
1,125397
+
+
-
-
-
-
+
+
8,003662
+
-
+
-
-
+
-
+
1,47084
+
+
+
-
+
-
-
-
9,316202
+
-
-
+
+
-
-
+
1,702583
+
+
-
+
-
+
-
-
5,99602
+
-
+
+
-
-
+
-
2,097892
+
+
+
+
+
+
+
+
3,4093
2,693845
Обработка результатов
Вычисляем коэффициенты регрессии.
b0 =
bi=
bij =
bijl =
и.т.д
Таблица № 17
Коэффициенты регрессии
b0
b1
b2
b3
b12
b13
b23
b123
4,140
2,541
-0,0667
-0,839
-0,252
-1,139
-0,481
-0,494
Проверяем значимость коэффициентов
Вычисляем дисперсию параметра оптимизации S2y
S2y=
Проверяем гипотезу о значимости коэффициентов регрессии по t – критерию Стьюдента. Для этого рассчитываем доверительный интервал для коэффициентов регрессии Δb
Δb=±t*Sb
Sb =
Таблица №18
Проверка значимости коэффициентов
основной уровень
Y1
Y ср
m-1
S^2
Sb^2
Sb
Δb
2,69384
2,51820
0,12897
0,01612
0,12697
0,35297
2,77937
2,18299
2,85426
2,08057
По результатам проверки получаем, что значимыми являются коэффициенты: b0,b1, b3,b13,b23,b123
Т.е. на параметр оптимизации будет влиять температура, ванна, температура-ванна, масса-ванна и сочетание всех трех факторов (температура, масса, ванна). Коэффициентb1положительный, поэтому, чем больше температура, тем лучше, коэффициент b3- отрицательный, поэтому лучше использовать ванну из никеля без добавления олова.
y=-10,766+2,42х1-0,4939х2-13,075х3+0,0988х1х2+2,662х1х3+0,4653х2х3-0,0988х1х2х3(в не кодированных)
Проверяем адекватность линейной модели.
Формальными признаками, по которым можно установить неадекватность линейной модели являются:
1. Незначимость хотя бы одного из эффектов взаимодействий
2. Незначимость суммы коэффициентов при квадратичных членах.
Оценкой этой суммы служит разность между b0 и значением параметра оптимизации в центре плана Ȳ0. Если эта разность превосходит ошибку опыта SȲ = , то линейная модель неадекватно описывает исследуемый процесс.
Ȳ-b0
SȲ
0,085583
0,032884
Линейная модель неадекватно описывает исследуемый процесс.
Литературный обзор.
Кислород, в зависимости от формы его нахождения в галогенидах, может быть определен различными методами.
Фторидный метод.
Основан на способности фтора замещать кислород в окислах металлов. Реакция фторирования протекает достаточно быстро (20-30 мин.) при температурах 300-4000С. В качестве реагентов, помимо газообразного фтора, могут применять вещества, содержащие высокоактивный фтор, такие как KBrF4 или CsBrF6. Замещение кислорода сопровождается его выделением в молекулярной форме:
2CaO+2СsBrF6→ 2CaF2 +O2↑+2CsBrF4
Объем выделившегося газа измеряют при атмосферном или пониженном давлении. С помощью этого метода возможно определение кислорода в любых галогенидах щелочных и щелочноземельных металлов, если при взаимодействии реактива с веществом не образуются летуче кислородные соединения, например оксигалогениды. Чувствительность определения зависит от чистоты реактива и количества кислорода, находящегося на стенках применяемой аппаратуры, и составляет 0,1-0,003 вес.%.
Метод изотопного разбавления.
При анализе данным методом не требуется полноты экстракции кислорода из пробы. Сущность метода состоит в следующем. Анализируемое вещество плавится в тигле вместе с образцом, содержащим известное количество кислорода, изотопный состав которого отличается от природного. Предполагается, что в течение некоторого промежутка времени изотопная концентрация кислорода во всем объеме печи выравнивается. Затем следует отбор газа и определение изотопного состава кислорода. Содержание кислорода в анализируемом образце рассчитывают по формуле:
Cx=
где Gd-количество кислорода, содержащегося в дозировочном образце
Чувствительность метода ограничивается из-за флуктуации величины поправки холостого опыта.
Физические свойства большинства галогенидов (температура плавления, летучесть, возможность восстановления окислов углеродом при температуре меньше 2500К) позволяют достаточно широко использовать метод изотопного разбавления. Однако наиболее целесообразно применение этого метода для анализа фторидов, в которых определение кислорода радиоактивационными методами затруднено вследствие присутствия фтора.
Метод радиоактивационного определения.
Для данного метода мог у т быть использованы многие ядерные реакции. Наибольшее применение получили следующие: на быстрых нейтронах -
016(n,р)N16; N16Т 1/2 - 7,35 сек;
на γ-квантах -
016(γ,n)015; 015 T 1/2 - 2,05 сек;
на заряженных частицах -
016(α, pn)F18;
O16(He3,p)P18;
O16(t,n)F18;
O16(p,n)F18;F18T 1/2 - 112,2 мин.
Щелочные, щелочноземельные металлы, хлор и йод не образуют при облучении изотопов, которые бы мешали определению кислорода. Бром при γ-активации образует изотоп с периодом полураспада, близким к 2 мин. Конечные продукты ядерных реакций кислорода и фтора с быстрыми нейтронами, заряженными частицами и γ-квантами идентичны. По этой причине раздельное определение кислорода и фтора таким методом невозможно. Исключение составляет определение кислорода во фториде лития с помощью облучения реакторными нейтронами по сопряженной реакции
Li6(n,α)t; Оl6(t,n)F18.
Ввиду малого сечения реакции F19(n,2n)F18 можно определять кислород во фториде лития с чувствительностью до 10-3%. Как правило, радиохимическое выделение не представляет самостоятельной задачи при проведении анализов радиоактивационными методами.
Метод активации быстрыми нейтронами, обладающий сравнительно невысокой чувствительностью10-3%, имеет преимущество перед другими радиоактивационными методами, заключающееся в том, что наведенную активность удается регистрировать прямыми инструментальными методами без радиохимического выделения изотопа. Чувствительность методов активации заряженными частицами составляет10-4-10-7%; γ-квантами 10-2-I0-4%.[1]
Масс-спектрометрия.
Применение масс-спектрального окончания приопределении ГП позволяет значительно снизить пределы обнаружения и повысить точность анализа. Благодаря разработке методики очистки поверхности образца перед анализом непосредственно в источнике ионов времяпролетного тандемного лазерного масс-рефлектронадостигнуты рекордные пределы обнаружения ГП для масс-спектральных методов анализа. Масс-спектральный метод пока является достаточно длительным, трудоемким и дорогим методом анализа.
ИК-спектроскопия.
Очень высокой чувствительностью обладают методы ИК-спектроскопии, применяемые при определении кислорода в кремнии и полупроводниковых материалах. Однако метод связан с использованием сложного оборудования, применим только для анализа твердых веществ, прозрачных в инфракрасном диапазоне излучения, и не всегда универсален из-за широкого набора спектров поглощения и отсутствия сведений о коэффициентах экстинкции.[2]
К методам определения общего содержания кислорода относятся высокотемпературное бромированиеKBrF4 и восстановительное плавление в присутствии углерода.
Достоинства восстановительного плавления: он проще, обладает большейвоспроизводимостью и позволяет использовать промышленную аппаратуру.
Метод основан на переводе кислорода образца в газовую фазу в форме окиси углерода при плавлении в вакууме или в среде инертного газа в графитовом тигле. Температура процесса восстановления может быть приблизительно определена на основании термодинамических расчетов.[1]
Для методов, используемых на практике для определения общего содержания кислорода, характерно следующее:
- среда в реакционной камере - вакуум или поток инертного газа (азот, аргон, гелий);
- графитовый тигель (одноразового применения) используется только для одной испытываемой навески или накопительный тигель (тигель многоразового использования) - для нескольких последовательно испытываемых навесок;
- реакционная среда - сухая (без жидкой фазы), т.е. в графитовый тигель помещают одну испытываемую навеску и восстановление проводят в твердофазном состоянии, т.к. анализируемый металл не плавится.
С целью ускорения восстановления некоторых металлов используют металлическую ванну (восстановление в жидкофазном состоянии). Для этого рекомендуется сначала приготовить ванну из плавкого металла (например платины, олова, железа, никеля), способного растворять как углерод, так и металл в испытываемой навеске. Нагрев непрерывный, если испытываемую навеску помещают в тигель, предварительно нагретый до температуры реакции; при этом восстановление происходит в несколько минут.
Нагрев импульсный, если холодный тигель с испытываемой навеской с помощью мощного импульса энергии нагревают за несколько секунд до пиковой температуры 3000 °С, при которой восстановление происходит очень быстро.
Определение кислорода по измерению содержания СО и СО2 осуществляют различными методами. Во всех методах используют устройства для химической конверсии, обеспечивающие полное превращение оксидов углерода в СО или СО2. Обычно используют следующие аналитические методы: объемный (для монооксида углерода); хроматографии (для монооксида углерода); инфракрасного поглощения (для диоксида и монооксида углерода); теплопроводности (для монооксида и диоксида углерода); кулонометрии (для диоксида углерода).
При непрерывном нагреве и восстановлении в твердом состоянии реакция восстановления может протекать медленно, и время полного восстановления оксидов зависит от содержания кислорода. Поэтому невозможно определить условия определения кислорода для каждого из анализируемых металлов, сплавов и карбидов и каждого типа применяемых приборов (установок).
Рекомендуется путем выполнения предварительных испытаний определять оптимальные условия анализа для конкретного типа материала и предполагаемого в нем содержания кислорода. С этой целью на одной и той же пробе проводят ряд последовательных испытаний, ускоряя процесс ее восстановления увеличением температуры и (или) времени реакции вплоть до момента, когда содержание измеряемого кислорода достигнет максимального и постоянного значения. При восстановлении можно также изменять другие параметры (например, металл ванны).
Для обеспечения установленных рабочих условий рекомендуется применять только, имеющие сертификаты, эталонные образцы (стандартные образцы по нормативным документам) того же типа, что и материал пробы. [3]
Особенности галогенидов щелочных и щелочноземельных металлов, как объектов анализа методом восстановительного плавления:
1. Упругость паров этих солей при температуре восстановления окислов соответствующих металлов велика, и потому их восстановительное плавление можно проводить только в атмосфере инертного газа.
2. Пары галогенидов весьма агрессивны и вызывают коррозию аппаратуры. Поэтому кварцевые детали печи следует охлаждать проточной водой.
3. В литературе нет сведений о поглощении выделенной из образца окиси углерода возгонами галогенидов или их парами. Испарение галогенидов (но не окислов металлов) практически не сказывается на результатах анализа, если оно не вызывает механического уноса кислородсодержащих примесей.
Применение этого метода ограничивается галогенидами, обладающими малой летучестью при температуре экстракции газов (1900-2000 К), такими, как фториды щелочноземельных металлов, хлориды кальция, стронция и бария, а также бромиды кальция и бария. [1]
При протекании реакции между различными оксидами с углеродом может образовываться не только СО, но и другие летучие соединения, которые затрудняют анализ.
a) 1/m MenOm + C→n/m Me + (CO)
b) 1/(m-n) MenOm+ C→n/(m-n) (MeO) + (CO)
c) Bi2O3, Cu2O, PbO, NiO,WO2, FeO, MoO2
MenOm + (CO)→n/m Me + (CO2)
d) Au2O3, Ag2O, или оксиды платиновой группы
2/m MenOm→2n/m Me + (O2)
e) Sb2O3, PbO, In2O3
MenOm→(MenOm).
Также существует проблема сосуществования СО и СО2 при определенных условиях. Это следует учитывать при определении точности в определении кислорода.[4]
Также на результаты анализа влияют сами графитовые тигли. Выделение кислорода в пористых тиглях протекает гладко с отличной повторяемостью, а в плотных тиглях кинетика реакции становится неустойчивой, происходит ухудшение пиков. Чтобы получить лучшие результаты очень важно, чтобы внутренние тигли были с пористой губчатой морфологией, которая предотвращает накопление газообразных продуктов реакции на поверхности раздела расплав / графита.[5]
Механизм восстановления можно представить состоящим из следующих стадий:
1) насыщение расплава углеродом: С(гр)→[C]
2) адсорбция атомов углерода на межфазной поверхности включение — расплав: [С]→Садс;
4) десорбция атомов R с поверхности включений в расплав:
5) образование пузырьков СО на оксидных частицах в расплаве:
СОадс→Сгаз;
6) флотация связки пузырек - включение на поверхность расплава, отрыв пузырьков от включений и переход в газовую фазу;
7) перенос газообразных продуктов реакции потоком гелия в газодинамической системе анализатора.[6]
Поверка газового анализатора HORIBA.
Определение СКО случайной составляющей погрешности измерения концентрации элемента осуществляется путем пятикратного измерения концентрации кислорода в государственных стандартных образцах.
По полученным результатам рассчитываем среднее арифметическое значение концентрации кислорода по формуле:
C =
Рассчитываем СКО случайной составляющей погрешности измерения концентрации кислорода по формуле:
СКО =
Рассчитываем относительное СКО случайной составляющей погрешности измерения концентрации кислорода по формуле:
СКО =
где n – число измерений
Ciи С – соответственно текущее и среднее арифметическое значение концентрации кислорода.
Анализатор признают годным для применения, если значение СКО случайной составляющей погрешности измерения концентрации кислорода не превышает 2% или значение относительного СКО случайной погрешности измерения концентрации кислорода не превышает 7%.


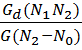
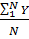
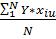



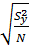
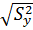 , то линейная модель неадекватно описывает исследуемый процесс.
, то линейная модель неадекватно описывает исследуемый процесс.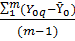
 C =
C = 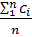
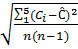
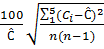
 где n – число измерений
где n – число измерений