

Историки об Елизавете Петровне: Елизавета попала между двумя встречными культурными течениями, воспитывалась среди новых европейских веяний и преданий...

Кормораздатчик мобильный электрифицированный: схема и процесс работы устройства...
Историки об Елизавете Петровне: Елизавета попала между двумя встречными культурными течениями, воспитывалась среди новых европейских веяний и преданий...
Кормораздатчик мобильный электрифицированный: схема и процесс работы устройства...
Топ:
Характеристика АТП и сварочно-жестяницкого участка: Транспорт в настоящее время является одной из важнейших отраслей народного...
Характеристика АТП и сварочно-жестяницкого участка: Транспорт в настоящее время является одной из важнейших отраслей народного хозяйства...
Когда производится ограждение поезда, остановившегося на перегоне: Во всех случаях немедленно должно быть ограждено место препятствия для движения поездов на смежном пути двухпутного...
Интересное:
Наиболее распространенные виды рака: Раковая опухоль — это самостоятельное новообразование, которое может возникнуть и от повышенного давления...
Искусственное повышение поверхности территории: Варианты искусственного повышения поверхности территории необходимо выбирать на основе анализа следующих характеристик защищаемой территории...
Принципы управления денежными потоками: одним из методов контроля за состоянием денежной наличности является...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Острый лимфобластный лейкоз (ОЛЛ) - тип лейкемии, чаще всего встречающийся у детей. Он исходит из недифференцирующегося пролиферирующего клона лимфобластов. Лейкозные лимфобласты вытесняют из костного мозга и замещают в крови миелоидные элементы.
Около 75% случаев ОЛЛ представлено пре-В-клонами (L1), до 20-25% - Т-клонами, относящимися к ранним тимоцитарным стадиям дифференцировки, подобными клеткам лимфобластной Т-лимфомы (L2), и лишь в нескольких процентах случаев (1-2%) неопластические клоны, сходные с клеточным субстратом лимфомы Бёркитта, представляют собой В - клетки (L3). Последний вариант рассматривается как лейкозная стадия течения этой лимфомы. Пик заболеваемости ОЛЛ приходится на возраст 4 - 5 лет, девочки составляют 1/3 больных. Преобладание больных мужского пола особенно характерно для ОЛЛ L2 (Т-клеточного). В связи с пролиферацией и расселением атипичных лимфобластов, больной ОЛЛ испытывает проявления панцитопении по миелоидным клеткам. Характерны анемия, тромбоцитопения, гранулоцитопения и соответствующие им клинические корреляты (геморрагический, гипоксический и инфекционно-септический синдромы). Присутствуют паранеопластические симптомы, связанные с цитокинами, производимыми клетками иммунной системы и лейкозными бластами (анорексия, исхудание, остеопороз и костные боли). В силу этого, продолжительность жизни больных ОЛЛ после установления диагноза полвека тому назад не превышала трёх месяцев. В настоящее время она, благодаря возросшей точности диагностики и эффективности терапии, составляет, в среднем, 4 года.
Из-за сходства клиники ОЛЛ и ОМЛ и внешней трудноотличимости лимфоидных и миелоидных бластов, диагностика базируется на лабораторных исследованиях генетических, иммунологических и цитохимических свойств лейкозных бластов (см. выше табл.??). Большое значение имеет положительная реакция на терминальную дезоксинуклеотидил-трансферазу у 95% лимфоидных бластов (у 80-95% миелоидных бластов она отрицательна). Но при фенотипе L3 лимфоидный клон происходит из более зрелых В-клеток, не имеющих данного энзима, подобно клеткам ХЛЛ и волосато-клеточного лимфолейкоза. Отрицательны цитохимические пробы на миелоидные лизосомальные ферменты и липиды. ШИК-реакция в половине лимфобластов даёт не присущее миелоидным клеткам глыбчатое распределение окраски.
|
|
Иммуноморфологически на клетках ОЛЛ выявляется маркер CD19, типичный для В-клеток. До разработки этой реакции не удавалось доказать В-принадлежность лимфобластов подтипа L1, в связи с чем в источниках до 1994 г. эта важнейшая форма ОЛЛ неверно считалась 0-лимфоцитарной (исходящей из "ни Т, ни В-клеток"). Поверхностные иммуноглобулины имеются лишь у более зрелых L3-В-бластов. Ниже приводится таблица, полезная для объективного распознавания различных лейкозов, в том числе - ОЛЛ и его подтипов - с помощью панели меченых моноклональных иммуноглобулинов против CD-маркеров. Данная процедура соответствует мировому стандарту дифференциальной диагностики лейкозов.
Табл. 15. CD-маркеры в дифференциальной диагностике лейкозов (по Шайнбергу - Гольде, 1994 - с изменениями и дополнениями).
| Маркер | Распределение | При каких лейкозах отмечается. |
| HLA-DR | Ранние миело- моноцитарные и В-клетки | ОЛЛ, ОМЛ (кроме М3), ХЛЛ, волосатоклеточный лейкоз |
| CD1 | Ранние Т | T-ОЛЛ |
| CD2 | Т | Т-ОЛЛ |
| CD3 | Зрелые Т | Т-ХЛЛ, ретровирусный Т-лимфолейкоз взрослых |
| CD5 | Т | Т-ОЛЛ; В-ХЛЛ |
| CD7 | Т | Т-ОЛЛ; 20% ОМЛ |
| СD10 (Common Acute Leukemiae Antigen = CALLA) | Пре-В, ранние Т | ОЛЛ L1, L2 |
| CD13 | Миеломоноцитарные | ОМЛ (все подтипы) |
| CD14 | Миеломоноцитарные | ОМЛ (чаще М4 и М5) |
| CD15 | Миеломоноцитарные | ОМЛ (наиболее дифференцированные подтипы) |
| CD16 | NK и гранулоциты | Лимфолейкоз или лимфомы из NK-клеток или Тg-лимфоцитов |
| CD19 | В | В-ОЛЛ, В-ХЛЛ и волосатоклеточный лейкоз |
| CD20 | В | В-ОЛЛ, В-ХЛЛ и волосатоклеточный лейкоз |
| CD21 | В-клетки промежуточных стадий дифференцировки | ХЛЛ |
| CD25 | Активированные В- и Т-лимфоциты | Волосатоклеточный лейкоз и ретровирусный Т-клеточный лейкоз взрослых |
| CD33 | Ранние миеломоноцитарные | Все подтипы ОМЛ |
| CD34 | Стволовые кроветворные клетки | ОЛЛ и ОМЛ М0-М2 |
| Поверхностные Ig | В-клетки промежуточных и конечных стадий дифференцировки | ОЛЛ L3, ХЛЛ и волосатоклеточный лейкоз. |
|
|
Несмотря на сходство клиники ОЛЛ и ОМЛ, некоторые проявления (лимфаденопатия, гепатоспленомегалия, менингеальные явления и нейролейкемия) более типичны для ОЛЛ, так как лимфобласты более активно расселяются, в то время как лейкостаз и окклюзионные нарушения чаще бывают при ОМЛ, возможно, из-за большего диаметра миелобластов. Хлоромы при ОЛЛ не встречаются. Отдельные подтипы ОЛЛ могут иметь клинические отличия. Так, при L2 и Т-клеточной природе лейкозного клона встречаются симптомы увеличения тимуса и лимфоузлов средостения, кроме того, данная форма имеет более позднее начало и часто бывает лейкемической, в то время, как остальные подтипы, наиболее вероятно, принимают сублейкемическое течение. Гиперурикемия, в наибольшей степени, свойственна ОЛЛ L3, когда бывают даже признаки "туморолитического синдрома" - с острым повышением содержания мочевой кислоты, лихорадкой и суставными болями. Форма L1 (при пре-В-лимфоцитарной природе бластов) имеет наилучший, а форма L3 - самый плохой прогноз. В первом случае 95% правильно лечимых больных могут рассчитывать на ремиссию и у 80-90% она оказывается стойкой (> 5 лет), во втором случае даже комплексная терапия приводит к ремиссии лишь в половине случаев, и притом характер ремиссии не такой стойкий. При Т-клеточной форме (L2) прогноз промежуточный, с вероятностью стойкой ремиссии не более 50%.
Важной в прогностическом отношении является цитогенетическая характеристика бластов при ОЛЛ, которая выявляет аномалии хромосом у 80% больных. Не более 5% больных детей, но больше трети взрослых пациентов с ОЛЛ имеют филадельфийскую хромосому (транслокацию 9 22). Таким образом, наличие этого признака не служит основой для дифференцирования ХМЛ от других лейкозов, как это считалось ранее! Впрочем, можно упомянуть, что и тени Клейн-Гумпрехта-Боткина, которые классически считаются признаком ХЛЛ (см. выше), изредка бывают и при ОЛЛ.
|
|
При наличии филадельфийской аномалии прогноз ОЛЛ у таких Ph+ - больных неизменно ухудшается. Рассматриваемая транслокация при ОЛЛ приводит к перемещению гена abl из 9-й хромосомы в точку разрыва кластерной области bcr (breakpoint claster region = bcr) хромосомы 22. Химерный белок bcr-abl - это онкопротеин-протеинкиназа, пострецепторный ростовой стимулятор. При ОЛЛ известна его p190-разновидность, при ХМЛ Ph+ отмечается иммунологически и структурно иной р210-вариант. При ОЛЛ подтипа L3 всегда бывает транслокация 8 14, с формированием химерного гена из энхансера тяжёлой цепи иммуноглобулинов и протоонкогена с-myc. При подтипе L1 (из пре-В - клеток) нередко бывает гипердиплоидия с числом хромосом от 51 до 60, и это коррелирует с хорошим прогнозом. Однако, если у таких пациентов L1 ОЛЛ протекает с транслокацией 1 19, а это бывает почти в каждом четвёртом случае данного подтипа ОЛЛ, - то прогноз ухудшается. Терапия ОЛЛ L1 и L2 основывается на применении комбинаций цитостатика (например, винкристина) и индуктора апоптоза лимфоцитов (например, преднизон). В качестве третьего компонента добавляют L-аспаргиназу. Особенностью терапии, по сравнению с лечением ОМЛ, является меньшая миелотоксичность этих средств. После вызова ремиссии практикуют консолидирующее лечение цитостатиками и антиметаболитами. Профилактике рецидивов способствуют интратекальная химиотерапия и иррадиация ЦНС при нейролейкемии, которая, в случае ОЛЛ, наступает примерно у половины больных. Так как Т-клетки имеют высокую активность аденозиндезаминазы, при L2 пытаются применять её блокатор пентостатин. Химиолечение и радиотерапия чреваты тяжёлыми ятрогенными осложнениями. Из-за облучения черепа и позвоночника нарушаются функция гипоталамо-гипофизарного аппарата, рост детей. Индуцируется вторичный смешанный иммунодефицит появляется высокий риск оппортунистической инфекции, чаще всего наступает стерильность. Возможны ятрогенные вторичные неопластические заболевания (ОМЛ, солидные опухоли), а у грудных детей интратекальная терапия и облучение мозга могут приводить к некротической лейкоэнцефалопатии. При ОЛЛ, обычно - при первом рецидиве, после достижения второй ремиссии эффективна пересадка аллогенного совместимого костного мозга или аутологичного костного мозга, собранного у самого пациента и сохраненного в период первой полной ремиссии, индуцированной химиопрепаратами. Имеется опыт применения иммунотерапии антителами против антигенов В-клонов при ОЛЛ. В настоящее время профессиональная психология медиков и кругозор пациентов перестраиваются, и ОЛЛ перестаёт восприниматься как абсолютно фатальное заболевание. С деонтологических позиций, важно помнить, что прогноз для разных его форм сильно отличается, и что, несмотря на оптимистические тенденции, излечимость большинства случаев ОЛЛ относительна и выражается не в достижении полного здоровья, а в возможности существенно продлить жизнь пациента ценой множества неизбежных вторичных ятрогенных нарушений.
|
|

|

|
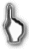
|

|

|
Редкие формы лимфолейкоза.
Волосатоклеточный лейкоз (ВКЛ) - редкое, но нозологически чётко очерченное заболевание. Это хронический лимфолейкоз взрослых, в основном - пожилых пациентов, при котором злокачественный клон исходит из В-лимфоцитов. Лейкозные элементы при ВКЛ имеют уникальные особенности:
· Несут, наряду с В-маркерами, еще и некоторые CD-антигены, характерные для Т-лимфоцитов и даже для моноцитов - в частности, СD11c и CD105 (см. табл.??);
· Имеют овоидное ядро, тонконитчатый хроматин и, под электронным микроскопом - мелковорсинчатую поверхность (откуда и название). В цитоплазме клеток электронная микроскопия выявляет особый спиралевидный органоид - пластинчатый рибосомальный комплекс.
· Содержат кислую фосфатазу, активную в присутствии солей виннокаменной кислоты (дают положительную пробу на тартрат-резистентную кислую фосфатазу).
Ретровирус - предполагаемый этиологический агент данной формы лейкоза, но однозначно это не доказано. Только при редких разновидностях ВКЛ, когда лейкозные клетки проявляют свойства Т-лимфоцитов, в них обнаружены копии генов ретровируса HLTV-II. Болезнь клинически протекает медленно, часто - в алейкемической форме, постепенно развиваются анемия, гранулоцитопения, тромбоцитопения, выраженная спленомегалия. Увеличение печени менее значительно, а лимфаденопатия вовсе нехарактерна. Без лечения срок жизни не превышает 2-3 лет, при лечении - более 4 лет. Лечение не всегда приводит к ремиссии, прогноз остается плохим, хотя применение спленэктомии и -интерферонотерапии его несколько улучшает.
Т-клеточный ретровирусный лимфолейкоз/лимфома взрослых (ТКЛЛВ) - эндемичная форма лимфолейкоза вирусной этиологии, подострого (изредка -острого и даже молниеносного) течения. Возбудитель - ретровирус HLTV-1.
|
|
Данное редкое заболевание привлекает внимание патологов, как единственная доказанно контагиозная человеческая неоплазия. Наблюдается ТКЛЛВ на острове Ямайка и других островах Карибского моря, в юго-восточной части США (отдельные случаи среди афро-американцев), на Окинаве и в юго-западной Японии. Вирус данного заболевания поражает CD4+ Т-лимфоциты. ДНК, комплементарная вирусной, выделяется из всех лимфоцитов лейкозного клона, но не из нормальных лимфоцитов того же пациента. Копия генома вируса встроена в одну и ту же хромосому и имеет у всех лейкозных Т-клеток сходную внутрихромосомальную локализацию, дополнительно подтверждая их клональность и инфицирование до момента трансформации. Лейкозные клетки больных выделяют вирус в культуральную жидкость, и последний может лишать здоровые Т-клетки способности к апоптозу. Доказано появление IgG против возбудителя у 90% больных ТКЛЛВ. Показана возможность передачи инфекции половым и трансмиссивным путём, а также трансплацентарно, с последующим вероятным (хотя и необязательным) развитием неоплазии. Патогенез ТКЛЛВ, очевидно, является многошаговым и требует дополнительных соматических мутаций в одном из поражённых вирусом клонов. Первый, вирусозависимый шаг - приобретение потенциального бессмертия, очевидно, связан с внесением в Т-клетку копий генов так называемого tat-региона вирусного генома. Данный регион содержит гены, усиливающие транскрипцию белков самого вируса и гены, которые заставляют пораженный Т-лимфоцит выделять ростовой фактор интерлейкин-2 (ИЛ-2), параллельно экспрессируя рецептор этого фактора. Перенос tat-последовательности трансгенным мышам вызывает у последних опухоли. На ранних стадиях трансформации патологический клон сам, аутокринно стимулирует собственную пролиферацию. На последующих стадиях новая соматическая мутация в этом клоне должна снять необходимость в ИЛ-2, делая рост лейкозного клона автономным. Многошаговый процесс требует большого латентного периода. От сероконверсии до первых признаков лейкоза проходит несколько лет или десятилетия. Вирус данного лейкоза/лимфомы, во многом, похож на вирус ВИЧ-инфекции. Клиника ТКЛЛВ включает лимфаденопатию, спленомегалию, гепатомегалию, паранеопластические явления, в частности, гиперкальциемию, а также лимфоцитоз в периферической крови. Бывает поражение кожи и прогрессирующая спастическая миелопатия, напоминающая поражение ЦНС при ВИЧ-инфекции. Неоплазма крайне злокачественна и приводит без лечения к гибели больного за 7-9 месяцев.
|
|
|
|
|
|
Лимфомы.
"Нигде в патологии хаос терминов так не затуманил ясные концепции - как в области лимфоидных опухолей"
Руперт Виллис (1948)
Лимфомы - неопластические заболевания из клонов лимфоцитов, формирующие солидные опухоли. В широком смысле, все лимфолейкозы могут быть включены в это определение на соответствующей стадии своего течения. Но, традиционно лимфомами считаются только такие лимфоидные неоплазмы, которые берут начало вне костного мозга (в лимфоузлах, в рассеянных неинкапсулированных скоплениях лимфоидной ткани слизистых оболочек, реже - в селезёнке и в тимусе). Принципиально важно, что из-за рециркулирующего характера нормальных лимфоцитов, лимфомы могут возникать во многих органах, не имеющих прямого отношения к кроветворной и иммунной системе, например, в яичнике - и некоторые из них даже проявляют тенденцию к первичной локализации вне лимфоидной ткани. О лейкозе говорят при костномозговом происхождении бластов и выраженной циркуляции их в крови. О лимфоме - при наличии локального признака "плюс-ткань". Так как к моменту установки диагноза при лимфолейкозе или лимфоме могут быть заселены бластами и костный мозг, и лимфоузлы - граница между лимфомами и лимфолейкозами становится относительной. Выше мы уже обращали внимание читателя на аналогию некоторых форм лимфолейкоза и определённых лимфом. Лимфомы могут прогрессировать в лимфолейкозную стадию и сосуществовать с лимфолейкозом.
В хирургии прежде лимфомой долгое время называли любую опухоль из клеток лимфоузла. Поэтому гистиоцитоз, берущий начало из макрофагальных клеток лимфатических узлов, известен под причудливым традиционным именем "гистиоцитарная лимфома" (см. выше - о терминах в онкогематологии). Помимо истинных гистиоцитарных опухолей лимфоузлов, "гистиоцитарными лимфомами" долго называли неоплазмы из похожих внешне на гистиоциты иммунобластов - то есть, зрелых Т- и В-лимфоцитов, претерпевших бласт-трансформацию и активацию. По современной номенклатуре, это имунобластные лимфомы. Наконец, лимфомой de facto является миеломная болезнь, поскольку при ней злокачественный клон исходит из лимфоидных плазматических клеток. К данной группе причисляется и лимфогранулёматоз, который Ходжкин (1832) описал именно как особую лимфому (лимфома Ходжкина). Однако, в лимфоидной природе клона злокачественных клеток при лимфогранулёматозе до недавнего времени существовали сомнения (см. ниже). Тем не менее, за океаном все остальные лимфомы именуют "неходжкинскими", и данный термин является, в отличие от многих в этой области, совершенно однозначным и навсегда истинным - ибо обозначает, что их описал не доктор Ходжкин, а другие специалисты.
Термин "лимфома" предложил в ХIХ cтолетии Бильрот. Ранее опухоли из лимфоцитов входили в группу, обозначавшуюся как "гематосаркомы".
Все лимфомы считаются злокачественными, но в различной степени. Поэтому среди принципов классификации этих опухолей имеется не только гистологический (по виду лимфоцитов и их диффузному, либо фолликулоподобному распределению) и иммуноморфологический (по их антигенным маркерам), но и онкологический - по малой, средней или высокой степени малигнизации. Причина столь особенной терминологии - тот факт, что все лимфоциты, а не только злокачественные лимфобласты, физиологически рециркулируют, выходят в ткани, возвращаются в лимфоидные органы, подвергаются "хоумингу". Поэтому, подходя к лимфомам со стандартной онкологической логикой, невозможно чётко отделить метастазирование от обычного поведения лимфоцитов.
Это еще раз доказывает справедливость предположения Бернета о том, что между возникновением развитой иммунной системы и учащением неоплазии в филогенезе есть закономерная связь (см. выше). В самом деле, иногда кажется, что быть нормальным лимфоцитом - это и есть та минимальная степень "недоброкачественного" поведения клетки, которую терпит многоклеточный организм: лимфоциты подвержены клональной экспансии, соматическому мутированию, расселению, они повреждают клетки хозяина, выделяют некоторые присущие и неоплазмам цитокины - чем не повод если не для зачисления в "плохие парни" - то для "постановки на учёт"?
По-видимому, поддерживать антигенный гомеостаз организма можно, только рискнув антигенным постоянством самих лимфоцитов. Не случайно экспрессия онкогенов в лимфоидных клонах так часто связана с мутациями именно в тех хромосомах, где хранятся коды иммуноглобулиновых цепей или Т-клеточного рецептора и где происходит физиологическая реаранжировка генов. Применяется несколько классификаций лимфом. Непротиворечивые классификации громоздки, а удобные - иммуногистологически противоречивы. На протяжении ХХ века несколько раз бывало так, что едва лишь онкологи создадут удовлетворительную классификацию лимфом - иммунологи меняют терминологию в области обозначения лимфоидных клеток, а то и сами концепции об их генезе. Примером может служить обессмысливание термина "ретикулосаркома", который обязателен для всех руководств, изданных в 50-е - 60-е годы (см. напр. Лавкович, Кржеминьска-Лавкович 1964). Дело в том, что ретикулярными клетками тогда называли поздние стадии дифференцировки еще не открытых В-лимфоцитов (иммунобласты), безосновательно приписывая им роль стволовых элементов для всех лимфоидных, а то и всех кроветворных клеток. Как уже упоминалось выше, с современных позиций, опухоли из этих клеток - иммунобластные В-лимфомы.
Первую классификацию лимфом на основе светомикроскопической оценки морфологии лимфоцитов и архитектуры ткани опухоли предложил в 1966 г. Раппопорт. По степени тканевого атипизма он выделил диффузный тип и фолликулярный тип лимфом. Последний воспроизводит в примитивном виде черты организации лимфоидных фолликулов и, что характерно - фолликулярные лимфомы имеют существенно лучший прогноз, по сравнению с несущими более выраженные признаки тканевого атипизма диффузными. По морфологии клеток классификация Раппопорта включает:
· хорошо дифференцированные лимфомы (клетки, явно похожие на типичный лимфоцит периферической крови, с правильным округлым ядром и небольшим количеством агранулярной цитоплазмы);
· плохо дифференцированные лимфомы (клетки имеют ядро неправильной или изогнутой формы).
· недифференцированные лимфомы (более крупные клетки, с избытком цитоплазмы, напоминающие макрофаги). В последней группе, по Раппопорту, оказываются "гистиоцитарные лимфомы" и смешанноклеточные лимфомы.
После открытия Т- и В-лимфоцитов и их разновидностей стало ясно, что классификация длимфом нуждается в пересмотре. Оказалось, что градации Раппопорта представлены иммунологически разнородными клетками, которые принадлежат к различным видам и стадиям дифференцировки лимфоцитов.
Так, хорошо дифференцированные лимфомы могли происходить из малых В- и Т-лимфоцитов, включая Т-эффекторы. Плохо дифференцированные лимфомы оказались разнородной группой, включающей опухоли из полустволовых лимфоидных ни-Т, ни-В-клеток, Т-лимфобластов и В-лимфобластов, а иногда - В-клеток фолликулярных центров. Последние клеточные элементы могли также представлять субстрат некоторых смешанных и даже псевдогистиоцитарных лимфом. "Гистиоцитарная" лимфома, вообще, распалась на иммуноморфологически различные немакрофагальные В- или Т-иммунобластные опухоли, и лишь небольшой процент случаев оказался представлен истинно гистиоцитарными клонами. Обобщая эти данные, с новыми классификациями лимфом выступили Льюкс и Коллинз (1974) в США и Леннерт (1973) в Европе. Принцип этих градаций был иммуногистологическим и соответствовал стадиям дифференцировки лимфоцитов и происхождению соответствующих лимфоматозных клонов.
Табл. 16. Классификация неходжкинских лимфом (По Льюксу-Коллинзу - Леннерту, 1974).
| Новообразование | Происхождение неопластического клона |
| 1.Из лимфоидных полустволовых клеток: | |
| Лимфобластная лимфома / ОЛЛ пре-В типа | Полустволовая лимфоидная клетка, лимфобласт; лишённый маркеров Т- и В- линий. |
| 2.Из клеток Т-лимфоидной линии: | |
| Лимфобластная Т-лимфома/ Т-клеточный ОЛЛ | Т-лимфобласт |
| Лимфоцитарная Т-лимфома/Т-клеточный ХЛЛ | Малый Т-лимфоцит |
| Иммунобластная Т-лимфома (иммунобластная лимфосаркома) | Бласт-трансформированный Т-лимфоцит (Т-иммунобласт) |
| Фунгоидный "микоз"/синдром Сезари | Посттимический CD4+ Т-лимфоцит |
| 3.Из клеток В-лимфоидной линии: | |
| Лимфобластная В-лимфома/В-клеточный ОЛЛ | В-лимфобласт |
| Лимфоцитарная В-лимфома/В-клеточный ХЛЛ | Малый В-лимфоцит |
| В-Лимфома мантийной зоны (из промежуточных клеток) | В-клетка мантийной зоны лимфоузла |
| Моноцитоидная В-лимфома | В-лимфоцит |
| Лимфомы из В-клеток фолликулярных центров, включая лимфому Бёркитта / ОЛЛ L3 | В-клетка фолликулярных центров |
| Иммунобластная В-лимфома (иммунобластная лимфосаркома) | В-иммунобласт |
| Плазмацитоидная лимфоцитарная В-лимфома (миелома) | Плазматическая В-клетка |
| 4. Из гистиоцитов лимфоидных органов: | |
| Истинная гистиоцитарная " лимфома" | Гистиоцит |
Позже, Национальный Институт Рака (NIC) США принял рабочую клиническую классификацию неходжкинских лимфом, основанную на прагматическом принципе хорошего, промежуточного или плохого прогноза ("классификация NIC", 1982). Все лимфомы, включённые в классификацию NIС, подразделяются на 3 группы.
· 1.Лимфомы низкой степени злокачественности - с длительным малосимптомным течением, лучшим прогнозом для жизни (в 60-70% случаев - более 5 лет), но высокой резистентностью к терапии. Сюда относят:
o 1.1. Лимфому (и ХЛЛ) из малых лимфоцитов - диффузную, хорошо дифференцированную.
o 1.2. Плазмацитоидную лимфому (при макроглобулинемии Вальденстрёма)
o 1.3. Фолликулярные лимфомы - из малых расщеплённных клеток и смешанно-клеточную.
o 1.4. В-лимфому мантийной зоны
o 1.5. Моноцитоидную В-лимфому
· 2.Лимфомы промежуточной степени злокачественности, которые имеют более агрессивное течение, но поддаются терапии, и при которых 35-45% пациентов живут дольше 5 лет, в том числе:
o 2.1. Фолликулярная крупноклеточная лимфома
o 2.2. Диффузная лимфома из малых расщеплённых клеток
o 2.3. Диффузная лимфома из крупных расщеплённых и нерасщеплённых клеток
o 2.4. Диффузная смешанно-клеточная лимфома
· 3. Лимфомы высокой степени злокачественности - все диффузные, агрессивные, без лечения быстро приводящие к летальному исходу, поддающиеся терапии при вероятности 5-летних ремиссий 23-32%, в том числе:
o 3.1.Иммунобластная лимфома
o 3.2.Лимфобластная лимфома
o 3.3.Лимфома из малых нерасщеплённых клеток, включая тип Бёркитта.
Нарекания в адрес классификации NIС были связаны с её неполнотой (не включены миеломная болезнь, синдром Сезари и другие нозологические формы) и с отстутсвием деления на Т- и В-лимфомы. Тем временем, как уже описывалось выше, большая часть случаев ОЛЛ L1 была переквалифицирована из "полустволовых лимфоидных 0-клеточных" (группа 1 в классификации Льюкса-Коллинза-Леннерта) в пре-В-клеточные. Требовалось привести в соответствие с этим и номенклатуру лимфом. Кроме того, появились представления о возможности неоплазм, исходящих из NK-клеток. Поэтому совершенствование класификаций было продолжено. Наконец, новым достижением в этом любимом жанре онкогематологов стала классификация Хэрриса и соавторов REAL (The Revisеd Euro-American Classification of Lymphoid Neoplasms), предложенная в 1995 г. Так как она наиболее непротиворечива и всеобъемлюща с патофизиологической точки зрения, знакомим с нею читателя.
Классификация REAL неходжкинских лимфом (по Хэррису и соавт., 1995).
· 1. В-клеточные:
o 1.1.Из предшественников В-клеток:
§ 1.1.1. Пре-В-клеточный лимфобластный лейкоз/лимфома
o 1.2. Из В-клеток периферического фенотипа:
§ 1.2.1. В-клеточный ХЛЛ/мелкоклеточная лимфоцитарная лимфома
§ 1.2.2. Лимфоплазмацитоидная лимфома/иммуноцитома
§ 1.2.3. Лимфома из мантийных клеток
§ 1.2.4. Лимфома из клеток фолликулярных центров, фолликулярная
§ 1.2.4.1. Из малых клеток; из больших клеток; смешанная - из малых и больших.
§ 1.2.4.2. Диффузная из малых клеток
§ 1.2.5. Лимфома из клеток маргинальной зоны
§ 1.2.5.1. Внеузловая (в неинкапсулированных лимфоидных элементах слизистых, с моноцитоидным или без моноцитоидного фенотипа)
§ 1.2.5.2. Узловая (в лимфоузлах, с моноцитоидным или без моноцитоидного фенотипа)
§ 1.2.5.3. Селезёночная (в селезёнке, с ворсинчатыми лимфоцитами или без)
§ 1.2.6. Волосатоклеточный хронический лимфолейкоз
§ 1.2.7. Плазмоцитома/миеломная болезнь
§ 1.2.8. Диффузная крупноклеточная В-лимфома
§ 1.2.9. Лимфома Бёркитта
· 2. Т-клеточные и NK-клеточные лимфомы:
o 2.1. Из предшественников Т-клеток:
§ 2.1.1. Т-лимфобластный лейкоз/лимфома
o 2.2. Т-клеточные лимфомы из клеток с периферическим фенотипом и NK-клеток
§ 2.2.1. Т-клеточный ХЛЛ/пролимфоцитарный лейкоз
§ 2.2.2. "Крупногранулярный" NK-клеточный и Т-клеточный лейкоз
§ 2.2.3. Грибовидный "микоз" и синдром Сезари
§ 2.2.4. Т-лимфома с периферическим фенотипом
§ 2.2.4.1. Смешанная лимфома, из средних и больших или больших клеток, лимфоэпителиоидная.
§ 2.2.5. Ангиоиммунобластная Т-лимфома
§ 2.2.6. Ангиоцентрическая Т-лимфома
§ 2.2.7. Кишечная Т-лимфома (с энтеропатией или без)
§ 2.2.8. Анапластическая крупноклеточная лимфома CD30+; Т-клеточная или 0-клеточная.
Мировая совокупная частота лимфом почти равна частоте лейкозов, эти заболевания представляют по 4% спектра онкологической патологии и вместе занимают шестую позицию среди всех злокачественных неопластических болезней по распространенности. Лимфомы, наряду с лимфобластным лейкозом, главная проблема детской онкогематологии и основная солидная опухоль у молодых людей. У детей часто встречаются фолликулярные лимфомы из малых нерасщеплённых лимфоцитов (50%) и лимфобластные лимфомы (40%), а также бывает (в основном, в старшем детском и подростковом возрасте) лимфогранулёматоз. У взрослых спектр лимфом более полиморфный, хотя чаще всего бывают фолликулярные мелкоклеточные расщеплённые формы (35%) и лимфогранулёматоз (15%). Лимфомы чаще всего поражают негроидов и реже всего - монголоидов. Пораженность европеоидов - промежуточная. Заболеваемость лимфомами в Америке и Африке выше, чем в странах Евразии. Так, в США заболеваемость лимфомами 13,2 на 100 000 населения в год, в России она намного ниже. Некоторые лимфомы эндемичны. Лимфома Бёркитта, весьма часто встречаемая в малярийном поясе, особенно - в Тропической Африке и на острове Папуа-Новая Гвинея, сравнительно редко наблюдается вне основного ареала. Ретровирусная Т-клеточная лимфома-лейкоз взрослых нередка на острове Окинава и на островах Карибского моря, а в других регионах представлена завозными случаями.
Этиология некоторых лимфом, вполне определённо - вирусная. Это доказано, в частности, для лимфомы Бёркитта (вирус Эпштейна-Барр), для человеческого ретровирусного Т-клеточного лейкоза/лимфомы (см. выше) и для некоторых других лимфом высокой степени злокачественности, а также для многих лимфом птиц и животных, включая приматов. Условия, повышающие риск возникновения лимфом, включают ряд факторов: наследственные и приобретённые иммунодефициты, аутоаллергическая болезни (целиакия, синдром Шёгрена, системная красная волчанка, аутоиммунный тиреоидит, дерматомиозит, ревматоидный артрит), действие некоторых лекарств (циклоспорины, фенитоин, цитостатики), облучение. В лимфоматозных клонах обычны соматические мутации и экспрессия протоонкогенов и химерных генов.
Отдельные наиболее распространённые формы лимфом имеют некоторые клинико-патофизиологические особенности. В-лимфомы, в целом, встречаются намного чаще Т-лимфом. Патогенетически важно, что в В-лимфомах часто присутствуют соматические мутации, вовлекающие хромосомы 14 (где находится энхансер тяжёлых цепей иммуноглобулинов), 2 (с локусами реаранжируемых k-лёгких цепей антител) или 22 (с локусами реаранжируемых l-лёгких цепей). В юкстапозиции с этими генами могут переноситься при транслокациях протоонкогены c-myc из 8-й или bcl-2 - из 18-й хромосом. Это ведет к гиперэкспрессии протоонкогенов, необходимому звену патогенеза лимфом. Наиболее часто наблюдаются опухоли из клеток фолликулярных центров лимфоузлов. Эти опухоли могут иметь диффузное строение, что связано с более плохим прогнозом и более выраженной злокачественностью. Однако, чаще всего, они сохраняют фолликулярную структуру, состоят из малых клеток с расщеплёнными ядрами и имеют благоприятное течение, с низкой скоростью обновления клеточного пула, причём поддаются химиотерапии алкилирующими цитостатиками и антиметаболитами. В лечении применяется трансплантация костного мозга. Клетки опухоли расселяются экстранодально, но пролиферируют медленно. Однако, как правило, эти лимфомы рецидивируют после химиотерапии и трансплантации. Наиболее характерная для данных лимфом мутация - транслокация t 14®18 bcl2. Диффузные крупноклеточные лимфомы характеризуются быстрым ростом, интенсивным обновлением клеточного субстрата и малой продолжительностью жизни пациентов, при отстуствии лечения. Они провоцируют ряд осложнений, в частности, задержку мочеиспускания, связанную с инфильтрацией стенок уретры и ее пережатием. Анапластические клетки данной разновидности лимфом чувствительны к химиотерапии, в частности - по наиболее распространённой схеме CHOP (циклофосфан, доксорубицин, винкристин, преднизон). Особенно способствуют благоприятному безрецидивному прогнозу молодой возраст пациента, низкая сывороточная активность ЛДГ и отстутствие внеузловых проявлений.
Лимфома Бёркитта - разновидность мелкоклеточной нерасщеплённой фолликулярной лимфомы, поражает, в основном, детей. Она представлена эндемической "африканской" формой и спорадической "американской". Африканская форма соответствует по распространенности ареалу малярии. При ней поражение касается, в первую очередь, верхней или нижней челюсти, гораздо реже - подчелюстных лимфоузлов, а экстранодальная диссеминация вовлекает скелет и, очень часто - ЦНС. При американской форме поражаются, вначале абдоминальные органы, причём, опять-таки, не сами лимфоузлы, а кишечник, яичник, забрюшинное пространство и т.д. Потом следует длительный рост опухоли на месте, а затем - распространение на кожу, кости, периферические лимфоузлы и, изредка - в ЦНС. Агрессивная терапия позволяет вызвать стойкую ремиссию почти у половины больных. В середине минувшего столетия эпидемиологию и клинику эндемической формы этой лимфомы изучил, доказав её связь с инфекцией, врач британской колониальной администрации Бёркитт, один из выдающихся подвижников медицины ХХ века. Интересно, что на всем протяжении своих многотрудных исследований, потребовавших разъездов по Тропической Африке, доктор Бёркитт работал, практически, в одиночку, а от государства получил только… дотацию в 50 фунтов стерлингов для покупки пишущей машинки и 200 фунтов на подержанный джип.
При африканской форме лимфомы Бёркитта доказана роль вируса Эпштейна-Барр, делающего клоны В-клеток бессмертными, а также необходимость нескольких дополнительных шагов онкогенеза, связанных с соматическими мутациями. На ранней стадии для формирующегося неопластического клона большое значение имеет продукция В-клеточного ростового фактора, стимулируемая малярией и другими хроническими инфекциями. Затем, транслокация с участием протоонкогена с-myc (8-я хромосома) и генов- энхансеров различных цепей иммуноглобулинов (хромосомы 14, 2 или 22) приводит к гиперэкспрессии химерного онкобелка и делает рост клона автономным от ростовых факторов. Для спорадической формы связь с вирусной и иной инфекцией чётко не прослеживается.
В-лимфомы мантийной зоны чаще поражают пожилых пациентов. Они отличаются массивной экстранодальной инфильтрацией органов и тканей, а также нередким наличием маркера CD5, несмотря на принадлежность к В-лимфомам. Многие из них имеют транслокацию 11®14 и химерные гены с участием bcl-1. Моноцитоидная В-лимфома - также заболевание пожилых, но экстранодальное вовлечение для неё не типично (не более, чем в 15% случаев). В-клетки этой лимфомы могут иметь сходство с моноцитами и моноцитарный маркер C11. Иммунобластные В-лимфомы клинико-эпидемиологически связаны часто с иммунопатологическими заболеваниями (иммунодефицитными состояниями, гиперчувствительностью, коллагенозами, тиреоидитом Хашимото, болезнью холодовой гемагглютинации). Они представлены крупными базофильными иммунобластами, часто - напоминающими плазматические клетки и являются быстро прогрессирующими, высоко злокачественными новообразованиями. Плазмоцитоидные В-лимфомы представлены клонами злокачественных плазматических клеток. Их ядра часто содержат особые образования - базофильные тельца Датчера. Характерной клинической особенностью данных лимфом служит парапротеинемия. Как правило, имеется аномальный избыток моноклональных IgM в сыворотке, придающий ей повышенную вязкость (макроглобулинемия Вальденстрёма). Последствиями могут быть протеинурия белками Бенс-Джонса и а
|
|
|

Архитектура электронного правительства: Единая архитектура – это методологический подход при создании системы управления государства, который строится...

Общие условия выбора системы дренажа: Система дренажа выбирается в зависимости от характера защищаемого...

История развития хранилищ для нефти: Первые склады нефти появились в XVII веке. Они представляли собой землянные ямы-амбара глубиной 4…5 м...
Адаптации растений и животных к жизни в горах: Большое значение для жизни организмов в горах имеют степень расчленения, крутизна и экспозиционные различия склонов...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!