

Общие условия выбора системы дренажа: Система дренажа выбирается в зависимости от характера защищаемого...

Археология об основании Рима: Новые раскопки проясняют и такой острый дискуссионный вопрос, как дата самого возникновения Рима...
Общие условия выбора системы дренажа: Система дренажа выбирается в зависимости от характера защищаемого...
Археология об основании Рима: Новые раскопки проясняют и такой острый дискуссионный вопрос, как дата самого возникновения Рима...
Топ:
Выпускная квалификационная работа: Основная часть ВКР, как правило, состоит из двух-трех глав, каждая из которых, в свою очередь...
Организация стока поверхностных вод: Наибольшее количество влаги на земном шаре испаряется с поверхности морей и океанов...
Определение места расположения распределительного центра: Фирма реализует продукцию на рынках сбыта и имеет постоянных поставщиков в разных регионах. Увеличение объема продаж...
Интересное:
Подходы к решению темы фильма: Существует три основных типа исторического фильма, имеющих между собой много общего...
Что нужно делать при лейкемии: Прежде всего, необходимо выяснить, не страдаете ли вы каким-либо душевным недугом...
Уполаживание и террасирование склонов: Если глубина оврага более 5 м необходимо устройство берм. Варианты использования оврагов для градостроительных целей...
Дисциплины:
|
из
5.00
|
Заказать работу |
Содержание книги
Поиск на нашем сайте
|
|
|
|
Предмет и задачи медицинской генетики
Медицинская генетика изучает роль наследственности в патологии человека, закономерности передачи от поколения к поколению наследственных болезней, разрабатывает методы диагностики, лечения и профилактики наследственной патологии, включая болезни с наследственной предрасположенностью. Результатом исследований в этом направлении становятся медицинские и генетические открытия и достижения, направленные на борьбу с болезнями и улучшение здоровья людей.
ЗАДАЧИ МЕДИЦИНСКОЙ ГЕНЕТИКИ
• Изучение наследственных болезней, закономерностей их наследования, особенностей патогенеза, лечения и профилактики;
• Изучение наследственного предрасположения и резистентности к наследственным болезням;
• Изучение патологической наследственности;
• Исследование теоретических медико-биологических проблем (биосинтез видоспецифических белков, синтез иммунных антител, генетические механизмы канцерогенеза);
• Изучение вопросов генной инженерии, разрабатывающей методы лечения наследственных болезней путем переноса генов нормального метаболизма в ДНК больного.
Клиническая генетика — прикладной раздел медицинской генетики. Ее достижения применяются для решения клинических проблем пациентов или их семей. Она дает ответ на вопросы: какая болезнь у пациента (диагноз), как ему помочь (лечение), как предупредить рождение больного потомства (прогноз и профилактика), как диагностировать и уменьшить вероятность развития болезни с наследственным предрасположением. В настоящее время в клинической генетике используются, с одной стороны, генетические методы (генетический анализ, молекулярно-биологические, цитогенетические, биохимические, иммуногенетические) и, с другой стороны, все современные методы клинического обследования: ультразвуковое исследование (УЗИ), магнитно-резонансная томография (МРТ), компьютерная томография (КТ), позитронно-эмиссионная томография (ПЭТ).
Хромосомная теория наследственности
Сформулирована в 1911 г. американским ученым Т. Морганом. Ее сущность заключается в следующем:
— основным материальным носителем наследственности являются хромосомы с локализованными в них генами; - гены наследственно дискретны, относительно стабильны, но при этом могут мутировать;
— гены в хромосомах расположены линейно, каждый ген имеет определенное место (локус) в хромосоме;
— гены, расположенные в одной хромосоме, образуют группу сцепления и наследуются совместно;
— число групп сцепления равно гаплоидному набору хромосом и постоянно для каждого вида организмов;
— сцепление генов может нарушаться в результате кроссинговера;
— частота кроссинговера прямо пропорциональна расстоянию между генами.
Значение этой теории заключается в том, что она дала объяснение законам Менделя, вскрыла цитологические основы наследования признаков и генетические основы теории естественного отбора.
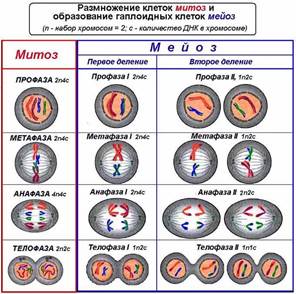
Митоз и мейоз
Митоз, или непрямое деление, наиболее широко распространен в природе. Митоз лежит в основе деления всех неполовых клеток (эпителиальных, мышечных, нервных, костных и др.). Митоз состоит из четырех последовательных фаз. Благодаря митозу обеспечивается равномерное распределение генетической информации родительской клетки между дочерними.
Период жизни клетки между двумя митозами называют интерфазой. Она в десятки раз продолжительнее митоза. В ней совершается ряд очень важных процессов, предшествующих делению клетки: синтезируются молекулы АТФ и белков, удваивается каждая хромосома, образуя две сестринские хроматиды, скрепленные общей центромерой, увеличивается число основных органоидов цитоплазмы.
В профазе спиралируются и вследствие этого утолщаются хромосомы, состоящие из двух сестринских хроматид, удерживаемых вместе центромерой. К концу профазы ядерная мембрана и ядрышки исчезают и хромосомы рассредоточиваются по всей клетке, центриоли отходят к полюсам и образуют веретено деления.
В метафазе происходит дальнейшая спирализация хромосом. В эту фазу они наиболее хорошо видны. Их центромеры располагаются по экватору. К ним прикрепляются нити веретена деления.
В анафазе центромеры делятся, сестринские хроматиды отделяются друг от друга и за счет сокращения нитей веретена отходят к противоположным полюсам клетки.
В телофазе цитоплазма делится, хромосомы раскручиваются, вновь образуются ядрышки и ядерные мембраны. В животных клетках цитоплазма перешнуровывается, в растительных — в центре материнской клетки образуется перегородка. Так из одной исходной клетки (материнской) образуются две новые дочерние.
Мейоз — это деление в зоне созревания половых клеток, сопровождающееся уменьшением числа хромосом вдвое. Он состоит из двух последовательно идущих делений, имеющих те же фазы, что и митоз.
| Фаза | Митоз | Мейоз | |
| I деление | II деление | ||
| Интерфаза | Набор хромосом 2n. Идет интенсивный синтез белков, АТФ и других органических веществ. Удваиваются хромосомы, каждая оказывается состоящей из двух сестринских хроматид, скрепленных общей центромерой. | Набор хромосом 2n Наблюдаются те же процессы, что и в митозе, но более продолжительна, особенно при образовании яйцеклеток. | Набор хромосом гаплоидный (n). Синтез органических веществ отсутствует. |
| Профаза | Непродолжительна, происходит спирализация хромосом, исчезают ядерная оболочка, ядрышко, образуется веретено деления. | Более длительна. В начале фазы те же процессы, что и в митозе. Кроме того, происходит конъюгация хромосом, при которой гомологичные хромосомы сближаются по всей длине и скручиваются. При этом может происходить обмен генетической информацией (перекрест хромосом) — кроссинговер. Затем хромосомы расходятся. | Короткая; те же процессы, что и в митозе, но при n хромосом. |
| Метафаза | Происходит дальнейшая спирализация хромосом, их центромеры располагаются по экватору. | Происходят процессы, аналогичные тем, что и в митозе. | Происходит то же, что и в митозе, но при n хромосом. |
| Анафаза | Центромеры, скрепляющие сестринские хроматиды, делятся, каждая из них становится новой хромосомой и отходит к противоположным полюсам. | Центромеры не делятся. К противоположным полюсам отходит одна из гомологичных хромосом, состоящая из двух хроматид, скрепленных общей центромерой. | Происходит то же, что и в митозе, но при n хромосом. |
| Телофаза | Делится цитоплазма, образуются две дочерние клетки, каждая с диплоидным набором хромосом. Исчезает веретено деления, формируются ядрышки. | Длится недолго Гомологичные хромосомы попадают в разные клетки с гаплоидным набором хромосом. Цитоплазма делится не всегда. | Делится цитоплазма. После двух мейотических делений образуется 4 клетки с гаплоидным набором хромосом. |
Моногенное наследование
Моногенным называется такой тип наследования, когда наследственный признак контролируется одним геном. Закономерности моногенной наследственности изучал выдающийся ученый Г. Мендель. Он экспериментально обосновал наличие единиц наследственности (наследственных задатков, наследственных факторов) и описал их основные свойства - дискретность, стабильность, специфичность аллельного состояния.
организмы в равной степени участвуют в передаче своих наследственных факторов потомкам.
Моногенные болезни наследуются в соответствии с законами классической генетики Менделя. Соответственно этому, для них генеалогическое исследование позволяет выявить один из трёх типов наследования: аутосомно-доминантный, аутосомно-рецессивный и сцепленное с полом наследование.
1)Аутосомно-доминантный тип наследования болезни имеет место в тех случаях, когда патологический ген является доминантным и обеспечивает развитие манифестной формы болезни даже в гетерозиготном состоянии, так как он локализуется на одной из двух гомологичных неполовых хромосом.
Данный тип наследования характеризуется следующими признаками:
• прямая передача болезни происходит от одного из родителей, что является прямой вертикальной передачей генетических признаков, в том числе от больного отца;
• нередко прослеживается манифестация болезни в нескольких поколениях.
Доминантные гены обладают различной пенетрантностью — вероятностью проявления действия мутантного гена у его носителя. При неполной пенетрантности мутантного гена отдельные члены семьи, имеющие мутантный ген, заведомо являющиеся носителями мутации (так называемые «облигатные» носители), могут на протяжении всей жизни оставаться клинически здоровыми, но при этом передать свой мутантный ген потомкам (детям). Аутосомно-доминантный тип наследования характерен для ряда заболеваний, таких как хорея Гентингтона, нейрофиброматоз, эссенциаль-ный тремор, торсионная дистония, различные формы наследственной дистонии и т. д.
2) Аутосомно-рецессивный тип наследования характеризуется:
1) проявлением заболевания у гомозигот по патологической мутации;
2) передачей заболевания от здоровых родителей детям с вероятностью 25%. При этом родители больных являются здоровыми гетерозиготными носителями мутаций в гене, и сегрегация их потомства в соответствии с менделевскими закономерностями составляет 1:2:1. В этом случае 25%-ый риск возникновения заболевания отражает вероятность для потомков унаследовать мутантный ген от обоих родителей.
В большинстве случаев, риск аутосомно-рецессивного заболевания в потомстве больного низкий, что связано с редкостью этих заболеваний и относительно невысокой частотой носительства соответствующих мутаций в популяции. Ситуация меняется, если больной вступает в брак с гетерозиготным носителем мутации. Риск заболевания у потомства в этом случае составляет 50% и не отличается от такового при аутосомно-доминантном наследовании, в связи с чем такое наследование иногда называют псевдодоминантным. Вероятность такого события значительно увеличивается при кровнородственном браке, когда оба супруга имеют определенную долю общих генов, полученных ими от общего предка.
Наиболее распространенные аутосомно-рецессивные заболевания — муковисцидоз, спинальная мышечная атрофия, а также некоторых заболеваний из группы нарушения половой дифференцировки.
Наследование, сцепленное с полом. Многие признаки у человека наследуются сцеплено с половыми хромосомами. Локализованные в половых хромосомах гены имеют свои особенности передачи в поколениях. Например, сцеплено с Х-хромосомой наследуются гемофилия, дальтонизм и др., с У-хромосомой — ген облысения, гипертрихоза и др.
У человека признаки, наследуемые через Y-хромосому, могут быть только у лиц мужского пола, а наследуемые через X-хромосому, - у лиц как одного, так и другого пола. Особь женского пола может быть как гомо-, так и гетерозиготной по генам, локализованным в X-хромосоме. А рецессивные аллели генов у нее проявляются только в гомозиготном состоянии. Поскольку у особей мужского пола только одна X-хромосома, все локализованные в ней гены, даже рецессивные, сразу же проявляются в фенотипе. Такой организм часто называют гомозиготным.
Синдром Марфана
У больных резко выражен астенический тип сложения (высокий рост, истончение подкожной клетчатки, мышечная слабость), удлинение пальцев кистей и стоп, долихоцефалия — изменение формы головы, когда продольный размер значительно превышает поперечный, так называемое птичье лицо — узкое, с близкорасположенными глазами, тонким носом и выступающей вперед верхней челюстью (прогнатия); деформация ушных раковин, высокое небо. Иногда наблюдается расщепление твердого неба (волчья пасть). Конечности, пальцы кистей и стоп удлинены, грудная клетка воронкообразной или килевидной формы, ребра тонкие и длинные, межреберные промежутки широкие, позвоночник искривлен.
Синдром Элерса — Данлоса
Вызывает недостаточное развитие коллагеновых структур в различных системах организма. Проявляется патологией кожи, опорно-двигательного аппарата, сердечно-сосудистой системы, глаз, гиперрастяжимостью кожи — взятая в складку кожа легко оттягивается. При этом оттянутая кожная складка быстро возвращается в исходное положение. Кожа тонкая, нежная, бархатистая на ощупь, слабо фиксирована с подлежащими тканями. Мышечно-скелетные включают деформации грудной клетки, кифоз, сколиоз, косолапость. Зубы у больных могут быть неправильно сформированы, аномально расположены, уменьшены в размерах или частично отсутствуют. Умственное развитие больных в большинстве случаев соответствует возрасту. Растяжение кожи.
Муковисцидоз
Характеризуется системным поражением экзокринных желез и проявляется тяжелыми расстройствами функций органов дыхания, желудочно-кишечного тракта и ряда других органов и систем. Частота муковисцидоза в по данным разных авторов, составляет от 1:2500 до 1:8000 новорожденных. Заболевание наследуется по аутосомно-рецессивному типу, характеризуется кистозным перерождением поджелудочной железы, желез кишечника и дыхательных путей из-за закупорки их выводных протоков вязким секретом; проявляется в форме хронической пневмонии, расстройств пищеварения.
Полигенное наследование
Полигенное наследование – наследование, при котором несколько генов определяют проявление одного признака.
Комплементарность — такое взаимодействие генов, при котором 2 или более генов вызывают развитие признака. Например, у человека гены, ответственные за синтез интерферона, располагаются на 2 и 5 хромосомах. Для того чтобы организм человека мог продуцировать интерферон, необходимо, чтобы хотя бы по одному доминантному аллелю присутствовало одновременно и на 2, и на 5 хромосоме. Обозначим гены, связанные с синтезом интерферона и располагающиеся на 2 хромосоме — А (а), а на 5 хромосоме — В (в). Варианты ААВВ, АаВВ, ААВв, АаВв будут соответствовать возможности выработки организмом интерферона, а варианты аавв, ААвв, ааВВ, Аавв, ааВв — неспособностью.
Тип наследования признаков, обусловленных действием многих генов, каждый из которых оказывает лишь слабое действие. Фенотипически проявление полигенно обусловленного признака зависит от условий внешней среды. У потомков наблюдается непрерывный ряд вариаций количественного проявления подобного признака, а не появление четко различающихся по фенотипу классов. В ряде случаев при блокировании отдельного гена признак не проявляется вообще, несмотря на его полигенную обусловленность. Это свидетельствует о пороговом проявлении признака.
Тип наследования признаков, обусловленных действием многих генов, каждый из которых оказывает лишь слабое действие. Фенотипически проявление полигенно обусловленного признака зависит от условий внешней среды. У потомков наблюдается непрерывный ряд вариаций количественного проявления подобного признака, а не появление четко различающихся по фенотипу классов. В ряде случаев при блокировании отдельного гена признак не проявляется вообще, несмотря на его полигенную обусловленность. Это свидетельствует о пороговом проявлении признака.
Так как на развитие полигенных признаков большое влияние оказывают факторы внешней среда, выявление роли генов в этих случаях затруднительно.

Полимерия — несколько генов действуют на один признак одинаково. При этом при формировании признака не важно, какой паре принадлежат доминантные аллели, важно их количество.
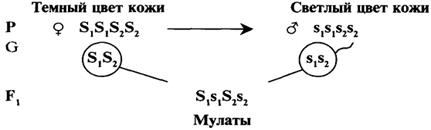
Например, на цвет кожи у человека влияет особое вещество — меланин, содержание которого обеспечивает палитру цвета от белого до чёрного (кроме рыжего). Наличие меланина зависит от 4-5 пар генов. Для упрощения задачи будем условно считать, что таких генов два. Тогда генотип негра можно записать — АААА, генотип белого — аааа. Светлокожие негры будут иметь генотип АААа, мулаты — ААаа, светлые мулаты — Аааа.

Плейотропия — влияние одного гена на появление нескольких признаков. Примером может служить аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлинённые конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечнососудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30-40 годами и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста. Среди известных исторических личностей данный синдром проявлялся у А. Линкольна, Н. Паганини, К.И. Чуковского (рис. 3.4, 3.5).
Эпистаз — подавление одним геном другого, неаллельного. Примером эпистаза может служить "бомбейский феномен". В Индии описаны семьи, в которой родители имели вторую (АО) и первую (00) группу крови, а их дети — четвёртую (АВ) и первую (00). Чтобы ребёнок в такой семье имел группу крови АВ, мать должна иметь группу крови В, но никак не О. Было выяснено, что в системе групп крови ABO имеются рецессивные гены-модификаторы, которые подавляют экспрессию антигенов на поверхность эритроцитов, и фенотипически у человека проявляется группа крови О.
Ещё одним примером эпистаза может служить появление белых альбиносов в семье темнокожих. В данном случае рецессивный ген подавляет выработку меланина, и если человек гомозиготен по этому гену, то независимо от того, какое количество доминантных генов, ответственных за синтез меланина, он имеет, окрас кожи у него будет альбиотический (рис. 3.6).
 |
 |
Синдром Морриса — синдром нечувствительности к андрогенам (синдром тестикулярной феминизации) проявляется нарушениями полового развития, которые развиваются в результате слабого реагирования на мужские половые гормоны у лиц с мужским набором хромосом (ХУ). Первым ввёл термин "синдром тестикулярной феминизации" американский гинеколог Джон Моррис в 1953 году.
Данный синдром является наиболее известной причиной развития мужчины как девушки или наличия проявлений феминизации у мальчиков, которые родились с мужским набором хромосом и нормальным уровнем половых гормонов. Имеются две формы андрогенной нечувствительности: полная или частичная нечувствительность. Дети с полной формой нечувствительности имеют однозначно женский внешний вид и развитие, в то время как люди с частичной формой могут иметь сочетание женских и мужских внешних половых признаков, в зависимости от степени нечувствительности андрогенов. Частота заболеваемости — примерно 1-5 на 100000 новорождённых. Чаще встречается синдром частичной нечувствительности к андрогенам. Полная нечувствительность к мужским половым гормонам является очень редким заболеванием.
Заболевание обусловлено мутацией в гене ЛЯ на Х-хромосоме. Этот ген определяет функцию андрогенных рецепторов — белка, который реагирует на сигналы от мужских половых гормонов и запускает клеточный ответ. При отсутствии активности андрогенных рецепторов не будет происходить развития мужских половых органов. Андрогенные рецепторы необходимы для развития лобковых и подмышечных волос, регулируют рост бороды и деятельность потовых желез. При полной андрогенной нечувствительности нет андрогенной активности рецепторов. Если некоторые клетки имеют нормальное количество активных рецепторов, то это синдром частичной нечувствительности к андрогенам.
Синдром наследуется с Х-хромосомой как рецессивный признак. Это означает, что мутация, вызывающая синдром, расположена на X-хромосоме. Согласно некоторым сведениям, в частности исследованию причин гениальности В.П. Эфроимсоном, синдром Морриса был у Жанны д'Арк.
Плейотропное действие генов
Плейотропное действие генов - это зависимость нескольких признаков от одного гена, то есть множественное действие одного гена.
У дрозофилы ген белого цвета глаз одновременно влияет на цвет тела, длины, крыльев, строение полового аппарата, снижает плодовитость, уменьшает продолжительность жизни. У человека известна наследственная болезнь - арахнодактилия ("паучьи пальцы"-очень тонкие и длинные пальцы), или болезнь Марфана. Ген, отвечающий за эту болезнь, вызывает нарушение развития соединительной ткани и одновременно влияет на развитие нескольких признаков: нарушение строения хрусталика глаза, аномалии в сердечно-сосудистой системе.
Классификация мутаций
1. По характеру проявления:
проявления бывают доминантными и рецессивными. Мутации нередко понижают жизнеспособность или плодовитость. Мутации, резко снижающие жизнеспособность, частично или полностью останавливающие развитие, называют полулетальными а несовместимые с жизнью — летальными.
2. По месту возникновения:
Мутация, возникшая в половых клетках, не влияет на признаки данного организма, а проявляется только в следующем поколении. Такие мутации называют генеративными. Если изменяются гены в соматических клетках, такие мутации проявляются у данного организма и не передаются потомству при половом размножении. Но при бесполом размножении, если организм развивается из клетки или группы клеток, имеющих изменившийся — мутировавший — ген, мутации могут передаваться потомству. Такие мутации называют соматическими.
3. По уровню возникновения:
Генные мутации – изменение строения одного гена. Это изменение в последовательности нуклеотидов: выпадение, вставка, замена и т.п. Например, замена А на Т. Причины – нарушения при удвоении (репликации) ДНК. Примеры: серповидноклеточная анемия, фенилкетонурия.
Хромосомные мутации – изменение строения хромосом: выпадение участка, удвоение участка, поворот участка на 180 градусов, перенос участка на другую (негомологичную) хромосому и т.п. Причины – нарушения при кроссинговере. Пример: синдром кошачьего крика.
Геномные мутации – изменение количества хромосом. Причины – нарушения при расхождении хромосом. В зависимости от характера изменения числа хромосом различают:
В зависимости от характера изменения числа хромосом различают:
Спонтанные мутации - возникают при нормальных условиях жизни, зависят от внешних и внутренних факторов, возникают в соматических и генеративных клетках.
Индуцированные мутации - это искусственное получение мутаций с помощью мутагенов различной природы. Впервые способность ионизирующих излучений вызывать мутации была обнаружена Г.А. Надсоном и Г.С. Филлиповым. В 1927 году американским ученым Джозефом Мюллером было доказано, что частота мутаций увеличивается с увеличением дозы воздействия. Ученые полагают, что факт наследования мутаций вызывает определенные опасения, поскольку это может увеличить риск развития рака. Азиатов от алкоголизма защищает ген-мутант. Почему процент алкоголиков в азиатских странах значительно ниже, чем в странах, где основную часть населения составляет так называемое белое население.
Факторы среды, вызывающие появление мутаций называются – мутагенами.
Различают:
Физические мутагены
- ионизирующее и ультрафиолетовое излучение;
- чрезмерно высокая или низкая температура;
Химические мутагены
- нитраты, нитриты, пестициды, никотин, метанол, бензпирен.
- некоторые пищевые добавки, например, ароматические углеводороды;
- продукты переработки нефти;
- органические растворители;
- лекарственные препараты, препараты ртути, иммунодепрессанты.
Биологические мутагены
- некоторые вирусы (вирус кори, краснухи, гриппа)
- продукты обмена веществ (продукты окисления липидов );
Свойства мутации:
Значение мутаций
Служат резервом наследственной изменчивости (сохраняются в популяции в скрытом-рецессивном) виде, являются материалом для эволюции.
Причина многих наследственных заболеваний и уродств.
Индуцированные мутации “поставляют” материал для искусственного отбора и селекции.
МУТАГЕНЕЗ - процессы-реакции в генном аппарате биологического объекта, при которых происходят изменения в строении генов, передающиеся по наследству. Такие изменения могут затрагивать отдельные нуклеотиды или группы их, сопровождаясь в некоторых случаях изменениями в морфологии хромосом. Изменения уже одного нуклеотида, входящего в состав триплета, приводят к образованию иной аминокислоты, входящей в состав белка, и могут привести к изменению соответствующего признака.
Мутагенез можно условно делить на спонтанный, когда мутации возникают в "нормальных" условиях роста, и индуцированный вследствие применения физических или химических мутагенов.
Спонтанный мутагенез зависит от внешних и внутренних факторов (биологические, химические, физические). Спонтанные мутации возникают у человека в соматических и генеративных тканях. Метод определения спонтанных мутаций основан на том, что у детей появляется доминантный признак, хотя у его родителей он отсутствует. При спонтанном мутагенезе могут происходить все типы наследственных перемен, которые наблюдаются при индуцированном мутагенезе: замена пар аденин-тимин или чаще гуанин-цитозин, ошибочное спаривание двух пуринов или двух пиримидинов, делеции, включения и другие изменения. Каждый биологический объект характеризуется определенным фоном спонтанных мутаций, которые с разной частотой затрагивают те или иные генетические признаки.
Индуцированный мутагенез - это искусственное получение мутаций с помощью мутагенов различной природы. Впервые способность ионизирующих излучений вызывать мутации была обнаружена Г.А. Надсоном и Г.С. Филлиповым. Затем, проводя обширные исследования, была установлена радиобиологическая зависимость мутаций. В 1927 году американским ученым Джозефом Мюллером было доказано, что частота мутаций увеличивается с увеличением дозы воздействия. В конце сороковых годов открыли существование мощных химических мутагенов, которые вызывали серьезные повреждения ДНК человека для целого ряда вирусов. Одним из примеров воздействия мутагенов на человека может служить эндомитоз - удвоение хромосом с последующим делением центромер, но без расхождения хромосом.
Хромосомные синдромы
Синдром Дауна
Кариотип 47 ХХ или 47 ХУ, 21+. Соотношение полов - МI: ЖI. Частота - 1: 700-800. характерная внешность: небольшая круглая голова со скошенным утолщенным затылком: монголоидный разрез глаз, эпикант, короткий седловидный нос, маленькие отстающие деформированные ушные раковины, полуоткрытый рот за счет макроглоссии, маленький западающий подбородок, своеобразная походка с неловкими движениями, косноязычие; - отставание в психомоторном развитии на первом году жизни; - слабоумие; - пороки развития сердечно-сосудистой системы (ДМЖП, ОАП); - пороки развития желудочно-кишечного тракта (атрезия пищевода); - склонность к инфекциям и злокачественным заболеваниям (лейкемия); - гипотрофия мышц, увеличение объема движений в суставах, поперечная ладонная складка; - пигментные пятна по краю радужки, косоглазие; - невысокий рост, гипотиреоз; - аномалии скелета: деформация грудины, укорочение и расширение кистей и стоп, клинодактилия и искривление мизинца, гипоплазия средней его фаланги, сандалевидная щель, может быть единственная складка на 5 пальце, готическое небо, мелкие зубы; - крипторхизм, гипоплазия полового члена.
Болезнь Шерешевского-Тернера.
Моносомия короткого плеча Х - хромосомы, синдром ХО. Кариотип 45 Х0. Болеют только женщины. Частота - 1: 10 000 новорож. девочек. Имеются три группы отклонений: 1) гипогонадизм (половой инфантилизм) выявляется в пубертатном периоде, аменорея в 96%, бесплодие - более 96-99%. 2) врожденные соматические пороки развития: - аномалии мочевой системы (подковообразная почка, удвоение почек и мочевыводящих путей) - 43-60% - умственная отсталость - 18-50% - аномалии сердечно-сосудистой системы (ВПР - коарктация) - 43% - нарушение слуха - 40-53% - нарушение зрения - 22% 3) низкий рост, при этом: короткое туловище - 97%, короткая шея - 71%, крыловидная складка на шее (птеригиум) - 53%, низкий рост волос на затылке - 73%.
Болезнь Клайнфельтера
Кариотип 47 ХХУ. Болеют только мужчины. Частота - 1:10 000 новорож. мальчиков. Клинические признаки заболевания проявляются в основном с наступлением пре- и пубертатного периода: - высокий рост - непропорционально длинные конечности (долихомелия) - гипоплазия яичек (99%) и полового члена (41%) - половой инфантилизм, нарушение сперматогенеза (100%), бесплодие - склонность к ожирению (по женскому типу), гинекомастия (55%) - снижение интеллекта, умственная отсталость (10%) - снижение полового влечения (70%)
Синдром полисомии по Х-хромосоме у женщины - “сверхженщина”
Кариотип 47, ХХХ. Болеют только женщины. Частота - 1: 1 000. Симптомы: - умственная отсталость различной степени в 75% - шизофрения с неблагоприятным типом течения. Больные с синдромами полисемии Х-хромосом обычно низкорослы. Аномалии ушей, прикуса, высокое (готическое) нёбо, короткие пальцы, короткий искривленный мизинец, широкое расстояние между I и II пальцами на стопах, неполная синдактилия и т.д. У некоторых больных могут быть пороки развития внутренних органов. Во многих случаях отмечается нормальное половое развитие. Иногда в период полового созревания обнаруживается недоразвитие вторичных половых признаков. Нарушение полового созревания особенно выражено при синдромах тетра- и пента — Х. При синдроме трисомии — Х возможны расстройства менструального цикла, однако большинство женщин способны к деторождению.
Генные болезни
Это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. Большинство генных патологий обусловлено мутациями в структурных генах, осуществляющих свою функцию через синтез полипептидов — белков. Любая мутация гена ведет к изменению структуры или количества белка.
мутантный аллель → измененный первичный продукт → цепь биохимических процессов в клетке → органы → организм
В результате мутации гена на молекулярном уровне возможны следующие варианты:
К генным болезням у человека относятся многочисленные болезни обмена веществ. Они могут быть связаны с нарушением обмена углеводов, липидов, стероидов, пуринов и пиримидинов, билирубина, металлов и др. Пока ещё нет единой классификации наследственных болезней обмена веществ.
Нарушения обмена углеводов
Пренатальная диагностика
В последние годы разработаны и внедрены методы, позволяющие установить наличие тяжелого дефекта плода ещё на начальных этапах беременности. Пренатальная диагностика показана 8% беременных.
Показаниями к проведению пренатальной диагностики являются:
1) Наличие в семье точно установленного наследственного заболевания;
2) Наличие предыдущего ребенка с множественными пороками развития, мертворождение или смерть предыдущего ребенка в младенчестве;
3) Возраст будущей матери более 35 лет, отца более 55 лет;
4) Близкородственные браки;
5) Длительная работа беременной на вредном производстве;
6) Повышенный уровень альфафетопротеина, хорионического гонадотропина, несвязанного эстриола в сыворотке крови беременной женщины.
В настоящее время с помощью пренатальной диагностики возможно выявление хромосомных болезней, а также более 100 ферментопатий.
УЗИ позволяет диагностировать тяжелые пороки развития скелета, установить пол плода (что важно при Х- сцепленном рецессивном типе наследования заболевания). Повторение УЗИ через каждые несколько недель может оказаться полезным для наблюдения за развитием плода.
Рентгенография плода используется только в случаях подоздрения на дефекты развития его костной системы. Применяется не ранее 30-й недели беременности.
Амниоцентез- извлечение околоплодной жидкости, содержащей клетки плода, - позволяет изучить хромосомный набор плода и выявить его нарушения. Биохимический анализ околоплодной жидкости позволяет диагностировать около 100 наследственных дефектов обмена веществ, дефекты незаращения нервной трубки. Амниоцентез проводится на 14-16 неделе беременности.
При необходимости диагностики в более ранние соки(7-10 недель) прибегают к хориоцентезу- взятию ворсинок эпителия хориона под контролем УЗИ для последующего цитогенетического, биохимического исследования и анализа ДНК.
Хорион- и амниоцентез являются инвазимными методами, которые в 1-3% случаев могут приводить к угрозе прерывания беременности.
Более безопасным методом является взятие крови плода из пуповины сосудов для диагностики хромосомных болезней и внутриутробных инфекций.
В последнее время диагностики некоторых наследственных болезней применяют методы анализа ДНК (генодиагностика). В России генодиагностика проводится в Москве, Санкт-Петербурге, Томске, Новосибирске, Уфе. Анализ ДНК определяет определить наличие не только патологических аллелей целого ряда моногенных заболеваний, но и идентифицировать ультрамалые кол-ва возбудителей микробных и вирусных инфекций, что дает возможность проводить доклиническую диагностику характера внутриутробного инфицирования плода.
Использования методов пренатальной диагностики является крупным шагом в профилактике наследственной и врожденной патологии.
Скрининг новорожденных
После рожден<
|
|
|
Организация стока поверхностных вод: Наибольшее количество влаги на земном шаре испаряется с поверхности морей и океанов (88‰)...

Кормораздатчик мобильный электрифицированный: схема и процесс работы устройства...

Особенности сооружения опор в сложных условиях: Сооружение ВЛ в районах с суровыми климатическими и тяжелыми геологическими условиями...

Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций...
© cyberpedia.su 2017-2026 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!

