Склонность гидрозолей к коагуляции под действием небольших добавок электролитов была замечена давно и послужила объектом большого числа экспериментальных и теоретических исследований.
Электролит, вызывающий нарушение агрегативной устойчивости золя, называется электролитом-коагулятором (или коагулянтом).
Коллоидные растворы очень чувствительны к присутствию посторонних электролитов и коагуляция может быть вызвана даже присутствием малых их количеств. Например, коагуляция наступает очень быстро, практически сразу же после образования золя, если он был получен в плохо вымытой посуде, сохранившей следы солей, содержащихся в водопроводной воде. Однако в присутствии каких-то определённых очень малых количеств электролита-коагулятора коллоидные растворы способны сохранять агрегативную устойчивость. Наименьшая концентрация электролита, вызывающая явную коагуляцию коллоидного раствора, называется порогом коагуляции g. Экспериментально порог коагуляции может быть определён постепенным (например, из бюретки) добавлением к исследуемому золю раствора электролита-коагулятора. Другой способ определения g - приготовление серии растворов электролита с равномерно увеличивающейся концентрацией с последующим добавлением к ним одинаковых объёмов золя. Наступление коагуляции может отмечаться визуально или с помощью приборов. Чаще всего для этих целей используются оптические методы. В любом случае порог коагуляции может быть вычислен по формуле
 ,
,
или, более точно (с учётом разбавления)
 ,
,
где С эк и V эк - соответственно концентрация и объём раствора элктролита-коагулятора, V золь – объём коллоидного раствора.
Порог коагуляции обычно измеряется в моль/л или в ммоль/л. При этом следует помнить, что физический смысл этой размерности – количество электролита (моль или ммоль), способное вызвать коагуляцию 1 литра коллоидного раствора.
Кроме порога коагуляции в коллоидной химии используется и обратная ему величина – коагулирующая способность Р:
 .
.
Размерность Р – л/моль или л/ммоль, что по физическому смыслу соответствует объёму золя, который может быть скоагулирован 1 молем (или миллимолем) электролита.
Порог коагуляции, а значит, и коагулирующая способность являются приблизительными характеристиками, так как зависят от очень многих факторов – от скорости прибавления электролита-коагулятора, от способа приготовления золя, от метода регистрации, от времени между добавлением электролита и моментом фиксирования явной коагуляции, от температуры и др.
Было обнаружено, что
коагулирующим действием обладают те ионы электролита-коагулятора, знак заряда которых противоположен знаку заряда гранул коллоидных мицелл, причём коагулирующее действие резко возрастает с увеличением заряда коагулирующего иона
(объединённое правило Г. Шульце ( 1882)- М. Гарди (1900)). То есть коагуляцию золя с отрицательно заряженными гранулами будут вызывать катионы, а золя с положительно заряженными гранулами – анионы.
При этом отношение порогов коагуляции одно-, двух- и трёхзарядных коагулирующих ионов приблизительно обратно пропорционально шестой степени их валентности:

Было также замечено, что вблизи порога коагуляции абсолютная величина z -потенциала, независимо от знака заряда гранулы, оказывается сниженной примерно до 25 - 30 мВ. Это его значение является критическим;при дальнейшем снижении величины z -потенциала золь практически полностью теряет устойчивость. Это является доказательством того, что главным в электростатическом факторе устойчивости золей является значение электрокинетического потенциала. Тщательные исследования коагулирующего действия различных ионов с одинаковой величиной заряда показали, что они образуют ряды, близкие к лиотропным рядам адсорбции.
Когда факторы устойчивости сняты не полностью, не каждое столкновение мицелл может закончиться их агрегированием. При этом в большинстве случаев для полной коагуляции золя требуется значительное время. Такая коагуляция называется медленной. Если же защитный фактор практически отсутствует, то мы имеем дело с быстрой коагуляцией,когда каждое столкновение частиц приводит к их объединению.
Следует отметить, что термины “быстрая” и “медленная коагуляция” относятся к механизму процесса, а не к реальному времени его протекания. Так, при значительных концентрациях мицелл “медленная” коагуляция может закончиться за короткое время из-за большой частоты столкновений, а в системах с малой концентрацией мицелл из-за малой частоты столкновений “быстрая” коагуляция может идти достаточно долго. Но в большинстве случаев реальная скорость коагуляции всё же прямо определяется механизмом.
Экспериментальное изучение кинетики коагуляции показывает, что быстрая коагуляция наблюдается при z -потенциале, равном нулю или при очень малых его значениях, намного более низких, чем критическое, а медленная - при значениях, близких к критическому.
Коагуляцией под влиянием электролитов объясняется бóльшая прозрачность морской и океанской воды по сравнению с речной, озёрной или болотной. В пресной воде, в особенности, в речной всегда имеется большое число дисперсий, в том числе и коллоидных частиц, которые образуются при размывании глин, глинистых грунтов и почв, а также при выветриваии других горных пород и т. д. Эти частицы придают воде мутность, иногда значительную. При впадении рек в моря эти дисперсии встречают электролиты – соли, содержащиеся в морской воде, и коагулируют под их действием. Образующийся осадок (коагулят) формирует около устий рек отмели, простирающиеся на большие расстояния, иногда на много километров или даже десятков километров. За пределами этих отмелей вода прозрачна, потому что не содержит дисперсий.
Другими жизненно важными примерами электролитной коагуляции могут служить образование холестериновых бляшек на внутренних поверхностях кровеносных сосудов или отложение солей в суставах, которое происходит при нарушениях солевого баланса плазмы крови. Сходный механизм приводит и к агглютинации эритроцитов при свёртывании крови.
6.5. Теории коагуляции
Общей количественной теорииагрегативной устойчивости и коагуляции пока не существует, хотя в процессе развития коллоидной химии возникло немало теорий, пытавшихся связать устойчивость гидрофобных золей и коагулирующее действие электролитов с теми или иными параметрами системы. Каждая из этих теорий объясняла ряд фактов, но оказывалась бессильной перед множеством других, поскольку эти теории, как правило, связывали сложный процесс коагуляции с каким-либо одним параметром системы.
Так, химическая теория П. Э. Дюкловыдвигала в качестве причины коагуляции химические реакции на границе раздела фаз, приводящие к нейтрализации поверхностного заряда.
Адсорбционная теория Г. Фрейндлихаобъясняла снижение заряда частиц адсорбцией ионов противоположного знака.
Согласно электростатической теории Г. Мюллераувеличение концентрации ионов электролита-коагулянта приводит при постоянном заряде на поверхности частиц к снижению их z -потенциала и, следовательно, к уменьшению устойчивости системы.
Теория А. И. Рабиновичарассматривала совместное действие ионного обмена и снижение z -потенциала. Роль z -потенциала в протекании коагуляции показана в п. 6.4. В теории В. Оствальдакоагуляция рассматривалась как вытеснение дисперсной фазы межионными силами притяжения, действующими в дисперсионной среде. В этом представлении параметром, определявшим коагуляцию, является коэффициент активности электролита.
По-видимому, все описанные в этих теориях механизмы коагуляции в той или иной степени имеют место. Преобладающее значение какой-либо из них приобретает в зависимости от природы дисперсной системы и коагулянта, а также от внешних факторов – температуры, механических воздействий и др.
В настоящее время широкое признание и распространение получила теория, основанная на физическом подходе к процессу коагуляции. В наиболее общем виде эта теория была разработана советскими учёными Б. В. Дерягиным и Л. Д. Ландау в 1937 - 41 гг. и несколько позднее (1948 г.) независимо от них голландскими учёными Э. Фервеем и Й. Овербеком. По первым буквам их фамилий эта теория названа теорией ДЛФО. Следует отметить, что, как и другие перечисленные ранее теории, теория ДЛФО не может претендовать на полное описание механизма коагуляции. Однако она дает возможность получения некоторых количественных зависимостей, например, позволяет рассчитывать величины порогов коагуляции для различных электролитов.
Теория ДЛФО основана на сопоставлении межмолекулярных взаимодействий частиц дисперсной фазы в дисперсионной среде, электростатического взаимодействия диффузных ионных слоёв и теплового броуновского движения частиц дисперсной фазы.
Эта теория на строгой количественной основе обобщила и развила представления об электростатической устойчивости золей, использованные в работах Г. Мюллера, А. И. Рабиновича, В. А. Каргина. Как говорилось выше, добавление электролита в коллоидную систему ведёт к сжатию двойного электрического слоя и, соответственно, к уменьшению расстояния, на котором между частицами действуют силы электростатического отталкивания. В таких условиях частицы, точнее, их твёрдые основы («агрегаты»), при броуновском движении могут подходить друг к другу на более близкие расстояния. На малых расстояниях резко возрастает роль сил межмолекулярного притяжения.
Таким образом, при сближении частиц на них действуют два вида
сил - силы притяжения и силы отталкивания. В соответствии с законом Ньютона силы притяжения убывают обратно пропорционально квадрату расстояния между частицами. Силы отталкивания - это с одной стороны электростатическое взаимодействие одинаково заряженных коллоидных частиц, а с другой - сопротивление структурно-механического слоя, имеющегося на поверхности частиц («расклинивающее давление» по Б. В. Дерягину). Действие расклинивающего давления, возникающего в перекрывавшихся гидратно-адсорбционных слоях при столкновении мицелл, накладывается на кулоновские силы, благодаря чему силы отталкивания находятся в сложной экспоненциальной зависимости от расстояния между частицами.
Таким образом, взаимодействие налетающих друг на друга коллоидных частиц будет определяться балансом положительной энергии отталкивания и отрицательной энергии притяжения. Его можно изобразить графически в виде энергетической кривой взаимодействия (рис. 6.1).
Видно, что на результирующей кривой имеется два минимума и один максимум. Их наличие говорит о том, что на малых и больших расстояниях между коллоидными частицами преобладает энергия притяжения, а на средних расстояниях - энергия отталкивания.
Рис. 6.1. Зависимость энергии взаимодействия сталкивающихся частиц
от расстояния d
(1- энергия электростатического отталкивания; 2 - энергия притяжения;
3 – результирующая энергия взаимодействия; min 1 – дальний минимум;
min 2 – ближний минимум; max – максимум)
Минимум min 1 отвечает притяжению частиц через прослойку среды. При сближении частиц до расстояния, соответствующего этому минимуму, они притягиваются друг к другу и собираются во флокулы. Небольшая глубина минимума обусловлена ослабленными из-за расстояния силами притяжения и действием расклинивающего давленияпрослойки среды. Поэтому такие флокулы сравнительно легко могут быть разрушены введением пептизатора, а часто и просто энергичным перемешиванием.
Максимум на средних расстояниях характеризует потенциальный барьер, препятствующий непосредственному касанию частиц. Он связан с возникновением расклинивающего давления перекрывающихся адсорбционно-сольватных слоёв и со значением электрокинетического потенциала z. Чем больше z -потенциал, тем более высоко проходит линия 1, и тем выше потенциальный барьер, не дающий частицам подойти слишком близко друг к другу. Практика показывает, что потенциальный барьер, обеспечивающий агрегативную устойчивость дисперсной системы, возникает уже при z -потенциале, равном 20 ¸ 30 мВ. По теории ДЛФО роль электролита-коагулятора сводится к уменьшению потенциального барьера за счёт электростатической составляющей, связанной с величиной z -потенциала. Количественные уравнения теории ДЛФО позволяют рассчитать соотношение порогов коагуляции золя различными электролитами. В соответствии с ними пороги коагуляции для одно-, двух- и трёхзарядных ионов должны относиться друг к другу обратно пропорционально шестой степени валентности ионов, что совпадает с соотношением, установленным правилом Шульце - Гарди.
Если потенциальный барьер невелик и энергия движения налетающих частиц превышает его, частицы могут сблизиться на расстояние, соответствующее минимуму min 2. Это сближение может привести к вытеснению гидратных оболочек и к непосредственному контакту. В случае твёрдых частиц это приводит к возникновению очень прочных связей и, как следствие, - к необратимой коагуляции, а в случае жидких или газовых частиц – к коалесценции (слиянию)
Теория ДЛФО носит приближённый характер, так как не учитывает многих факторов коагуляции, например, таких, как природа ионов с одинаковым зарядом, выражаемая лиотропными рядами коагуляции, специфическая адсорбция ионов и др.
6.6. Кинетика коагуляции
Основы учения о кинетике коагуляции, т. е. о её протекании во времени, были заложены в начале ХХ века М. Смолуховским и затем развиты Г. Мюллером, Н. А. Фуксом и другими исследователями.
В основе теории Смолуховского лежит представление о том, что при нарушении агрегативной устойчивости происходит слияние только двух частиц. Вероятность одновременной встречи в пространстве трёх или более частиц чрезвычайно мала, поэтому механизм коагуляции можно представить таким образом. При столкновении двух одиночных частиц возникает двойная частица, при столкновении двойной и одиночной – тройная, двух двойных – четверная и т. д. Таким образом, коагуляцию можно формально рассматривать как бимолекулярную химическую реакцию. Скорость коагуляции будет определяться концентрацией частиц в объёме системы и скоростью диффузии частиц, которая, в свою очередь зависит от вязкости среды и температуры. Теория позволила вывести уравнение, связывающее концентрацию укрупнённых (агрегированных) частиц n а, образовавшихся за время t, с начальной концентрацией одиночных частиц n 0:
 , (6.1)
, (6.1)
где К – константа скорости коагуляции.
Поскольку константу скорости коагуляции трудно определить экспериментально или вычислить, можно использовать уравнение, включающее в себя время половинной коагуляции t 1\2, за которое число частиц уменьшается вдвое. Из уравнения (6.1) следует, что
n 0/2 = 1 + Kn 0 t 1/2,
и значит
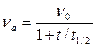 .
.
Время половинной коагуляции с учётом уравнений Фика для скорости диффузии и Эйнштейна для коэффициента диффузии можно вычислить по уравнению
 ,
,
где h - вязкость дисперсионной среды, k – константа Больцмана, T – абсолютная температура.