Нейрофиброматоз I типа является заболеванием с неясным прогнозом, потому что не существует четких критериев, позволяющих достоверно предсказать течение заболевания. Какого размера будут опухоли, где они будут располагаться, как поведут себя в течение жизни, возникнет ли рецидив удаленной опухоли – на эти вопросы невозможно ответить. Достоверно известно только, что беременность провоцирует увеличение количества нейрофибром.
Доброкачественные опухоли не несут в себе риск для жизни, однако если процесс переходит в злокачественный, тогда прогноз ухудшается.
Срок жизни больных с нейрофиброматозом I типа, в среднем, соответствует таковому в популяции (за исключением случаев перерождения опухолей в злокачественные).
Таким образом, болезнь Реклингхаузена представляет собой распространенную генетическую проблему человечества. Это заболевание с многоликой клинической картиной, в которой наиболее типичным является поражение кожи, нервной системы с развитием множества опухолей. Болезнь передается по наследству. Основным методом лечения в настоящее время является хирургический метод, однако ведутся исследования ряда препаратов, которые смогут помочь таким больным более эффективно.
Cиндром Марфана (Болезнь Марфана)
Синдром Марфана (англ. Marfan syndrome, болезнь Марфана) - аутосомно-доминантное генетическое заболевание которое поражает соединительную ткань, характеризующееся диспропорционально длинными конечностями, тонкими худыми пальцами, соответственно худым телосложением и наличием сердечно-сосудистых пороков, которые специфически проявляются в виде пороков сердечных клапанов и аорты. Это генетическое заболевание связано с нарушением функционирования соединительной ткани и значительным полиморфизмом клинических проявлений.
Это генетическое заболевание связано с нарушением функционирования соединительной ткани и значительным полиморфизмом клинических проявлений.
Преимущественно эта болезнь наследуется по доминантному признаку и вызывается аномалией гена FBN1, кодирующего белок фибрилин-1. У каждой личности есть пара таких генов. Поскольку наследование происходит по доминантному типу, то люди, что наследуют один аномальный ген FBN1 от кого либо из родителей будут поражены указанным заболеванием. Синдром Марфана может появляться в умеренной и тяжелой формах. Люди с этим заболеванием, как правило, высокие, с длинными конечностями и длинными худыми пальцами. Наиболее серьезными осложнениями болезни является повреждение клапанов сердца и нарушение структуры стенок аорты. Также заболевание может влиять на легкие, глаза, твердую оболочку спинного мозга, скелет и твердое нёбо.
Кроме функций связующего белка, который служит опорой для ткани за пределами клетки, белок фибрилин связывается с другим белком, вследствие чего образуется трансформирующий фактор роста бета (TGF-β). TGF-β имеет негативное влияние на сосудистый тонус гладких мышц и нарушает развитие целостного внеклеточного матрикса. Еще одной причиной развития болезни вследствие мутации гена ответственного за синтез фибрилина, сегодня ученые называют накопление избыточного количества TGF-β в легких, клапанах сердца и в аорте, что ослабляет ткани и вызывает симптомы болезни Марфана. Поскольку блокаторы рецепторов ангиотензина уменьшают количество TGF-β, что было продемонстрировано с помощью фармакологических средств, блокирующих функцию этих рецепторов (напр. лозартан и др.). В небольшом клиническом исследовании с участием молодых людей с тяжелой формой болезни Марфана, применявшие блокаторы рецепторов к ангиотензину II у некоторых пациентов действительности рост аорты существенно сократился, и соответственно риск тяжелых сердечнососудистых осложнений снизился.
Болезнь получила название от имени Антуана Марфана, французского педиатра, который впервые описал симптомы заболевания в 1896 году, заметив черты синдрома у пятилетней девочки. Ген, который вызывает болезни был впервые обнаружен Франческо Рамиресом в центре Маунт Синай (Нью-Йорк) в 1991 г.
Симптомы
Хотя нет никаких уникальных симптомов болезни Марфана, однако, сочетание таких признаков как длинные конечности, дислокация хрусталика, аневризма корня аорты, вполне достаточно для того, чтобы с уверенностью поставить диагноз. Однако, более чем 30 других клинических признаков, связанные с исследуемым синдромом. Большинство из них характеризуются изменениями скелета, кожи и суставов. Существует также сравнительно высокая вероятность того, что в разных семьях наблюдаться идентичные мутации.
Поражение костной системы
Большинство в 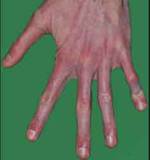 идимых признаков болезни Марфана, связанные с костной системой. У многих людей с синдромом Марфана рост значительно выше среднего. Некоторые из них имеют длинные конечности с длинными тонкими пальцами рук и ног (арахнодактилия ). Кроме диспропорций развития конечностей и чрезмерного роста болезнь Марфана вызывает и другие нарушения в функционировании костной системы. Особенно распространены искривление позвоночника (сколиоз), воронкообразная (внутрь) и килевидная (наружу) деформация грудной клетки, чрезмерная гибкость суставов, высокое небо, неправильный прикус, плоскостопие, молоткообразная деформация пальцев стопы (когда суставы пальцев на ногах согнуты и напоминают, поэтому молоток, сутулость, появление беспричинных растяжек на коже (стрии). У некоторых пациентов может появляться боль в суставах, костях и мышцах. Также у больных с синдромом Марфана иногда возникают расстройства или нарушения речи (через высокое небо и малые размеры челюсти). Ещё существует вероятность развития остеоартрита в раннем возрасте.
идимых признаков болезни Марфана, связанные с костной системой. У многих людей с синдромом Марфана рост значительно выше среднего. Некоторые из них имеют длинные конечности с длинными тонкими пальцами рук и ног (арахнодактилия ). Кроме диспропорций развития конечностей и чрезмерного роста болезнь Марфана вызывает и другие нарушения в функционировании костной системы. Особенно распространены искривление позвоночника (сколиоз), воронкообразная (внутрь) и килевидная (наружу) деформация грудной клетки, чрезмерная гибкость суставов, высокое небо, неправильный прикус, плоскостопие, молоткообразная деформация пальцев стопы (когда суставы пальцев на ногах согнуты и напоминают, поэтому молоток, сутулость, появление беспричинных растяжек на коже (стрии). У некоторых пациентов может появляться боль в суставах, костях и мышцах. Также у больных с синдромом Марфана иногда возникают расстройства или нарушения речи (через высокое небо и малые размеры челюсти). Ещё существует вероятность развития остеоартрита в раннем возрасте.
Нарушение функций глаз и зрения
Болезнь Марфана может влиять на зрение и глаза. Обычно у пациентов наблюдаются астигматизм и близорукость, однако 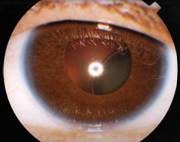 иногда фиксируется и дальнозоркость. Нарушение положения хрусталика в одном или обоих глазах (эктопия хрусталика), наблюдается у 80% больных. Выявить эти проблемы со здоровьем может офтальмолог (окулист) с помощью щелевой лампы (метод биомикроскопии). При синдроме Марфана дислокация преимущественно имеет суперотемпоральний характер (вверх и наружу), тогда как при подобном состоянии гомоцистеинемии - инфероназальний (вниз и внутрь). Иногда проблемы со зрением возникают только после ослабления соединительной ткани, которое вызвано расслоением сетчатки. Еще одной из офтальмологических проблем, связанных с синдромом Марфана можно назвать раннюю (в молодом возрасте) глаукому.
иногда фиксируется и дальнозоркость. Нарушение положения хрусталика в одном или обоих глазах (эктопия хрусталика), наблюдается у 80% больных. Выявить эти проблемы со здоровьем может офтальмолог (окулист) с помощью щелевой лампы (метод биомикроскопии). При синдроме Марфана дислокация преимущественно имеет суперотемпоральний характер (вверх и наружу), тогда как при подобном состоянии гомоцистеинемии - инфероназальний (вниз и внутрь). Иногда проблемы со зрением возникают только после ослабления соединительной ткани, которое вызвано расслоением сетчатки. Еще одной из офтальмологических проблем, связанных с синдромом Марфана можно назвать раннюю (в молодом возрасте) глаукому.
Система кровообращения
Наиболее серьезными признаками и симптомами болезни Марфана, является нарушение деятельности сердечно-сосудистой системы. Чрезмерная усталость, отдышка, нарушения ритма сердца, тахикардия (учащенное сердцебиение), стенокардия (которая сопровождается возникновением болевых ощущений в спине, плече или руке) - это те нарушения, которые наблюдаются при синдроме Марфана. Часто у пациентов, из-за нарушения кровообращения конечности (руки, ноги) холодные. Причинами для дальнейших обследований такого пациента может быть присутствие шумов в сердце, изменения на ЭКГ (электрокардиограмма) или присутствие симптомов стенокардии. Одной из причин регургитации (движения крови в противоположную сторону нормального направления), которая возникает из-за пролапса аортального или митрального клапанов сердца, можно назвать кистозную медиальную дегенерацию клапанов, которая возникает из-за влияния болезни Марфана.
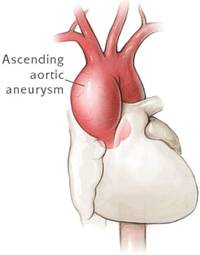 Однако основным признаком для дальнейшего детального исследования и изучения заболевания является расширенная аорта или аневризма аорты (выпячивание стенки аорты). Однако, иногда, очевидных проблем с сердечной системой не наблюдается, но ослабление соединительной ткани (через кистозную дегенерацию средней стенки сосудов - медии) вызывает аневризму или расслоения восходящей части аорты, что требует хирургического лечения. Расслоение аорты часто сопровождается болями в спине или груди и приводит к возникновению ощущения надрыва. Через нарушение функциональности соединительной ткани (что является патогенетическим механизмом развития синдрома Марфана) увеличивается частота случаев, при которых искусственный митральный клапан не приживается в организме больного. Именно поэтому необходимо проявлять чрезвычайную осторожность при лечении клапанов сердца. Предпочтение следует отдавать таким мерам, которые направлены на восстановление функциональности больного клапана, а не на немедленную его замену.
Однако основным признаком для дальнейшего детального исследования и изучения заболевания является расширенная аорта или аневризма аорты (выпячивание стенки аорты). Однако, иногда, очевидных проблем с сердечной системой не наблюдается, но ослабление соединительной ткани (через кистозную дегенерацию средней стенки сосудов - медии) вызывает аневризму или расслоения восходящей части аорты, что требует хирургического лечения. Расслоение аорты часто сопровождается болями в спине или груди и приводит к возникновению ощущения надрыва. Через нарушение функциональности соединительной ткани (что является патогенетическим механизмом развития синдрома Марфана) увеличивается частота случаев, при которых искусственный митральный клапан не приживается в организме больного. Именно поэтому необходимо проявлять чрезвычайную осторожность при лечении клапанов сердца. Предпочтение следует отдавать таким мерам, которые направлены на восстановление функциональности больного клапана, а не на немедленную его замену.
Во время беременности у женщин с синдромом Марфана, даже при отсутствии видимых отклонений в работе сердечно-сосудистой системы, высокий риск расслоения аорты, что может привести к летальному исходу, даже при своевременном лечении. Именно поэтому, если у женщины присутствует это заболевание, то перед зачатием необходимо пройти тщательное исследование и получить консультацию врача. А во время самой беременности каждые шесть-десять недель нужно проводить эхокардиографию для определения диаметра корня аорты. Во многих случаях возможны естественные роды без осложнений, однако, лишь после исчерпывающего медицинского обследования и оценки всех возможных рисков.
Влияние на лёгкие
Болезнь Марфана является одним из факторов риска для спонтанного возникновения пневмоторакса, при котором воздух выходит из легких и занимает плевральную полость между грудной клеткой и легкими, в результате чего легкие сжимаются. Больной пневмотораксом испытывает резкую боль в груди, дышит часто и поверхностно, наблюдается выраженная отдышка. Часто проявляется бледность или синюшность кожных покровов, в частности лица (цианоз). Если заболевание не лечить, оно может привести к смерти больного.
Кроме этого, синдром Марфана может быть связан с такими заболеваниями легких как апноэ во сне (это прекращение вентиляции легких во время сна, более чем на 10 секунд) и другими идиопатическими (с неустановленной причиной) обструктивными болезнями легких.
Влияние на центральную нервную систему
Одним из последствий болезни Марфана, который может негативно повлиять на качест  во жизни человека (хотя он не представляет угрозы жизни) является дуральная ектазия. Это ослабление и растяжение твердой оболочки мозга, а точнее соединительной ткани дурального мешка - мембраны, которая окутывает спинной мозг.
во жизни человека (хотя он не представляет угрозы жизни) является дуральная ектазия. Это ослабление и растяжение твердой оболочки мозга, а точнее соединительной ткани дурального мешка - мембраны, которая окутывает спинной мозг.
В течение длительного времени симптомы дуальной ектазии (боль в пояснице, в ногах, в области живота и таза, другие неврологические симптомы в нижних конечностях или головная боль) могут не проявляться. Или же резко исчезают, когда человек лежит на плоской ровной поверхности, на спине. При болях такого типа врачи обычно назначают рентген поясничного отдела позвоночника, хотя, как правило, дуральную ектазию невозможно заметить с помощью рентгена на ранних стадиях. Именно поэтому, ухудшение симптомов и отсутствие, какой либо другой причины боли создает необходимость проведения исследования с помощью магнитно резонансной томографии (МРТ) поясничного и крестцового отделов позвоночника. Дуральную ектазию, которая вызывает такие симптомы, будет хорошо видно на вертикальном изображении МРТ. Она будет иметь вид расширенных отростков, которые направлены к поясничным позвонкам. Другие неврологические проблемы, связанные с синдромом Марфана - это дегенеративные заболевания междупозвоночных дисков и костей спины. Также синдром Марфана является важным фактором вызывающим развитие дисфункции автономной нервной системы.
Патогенез заболевания
Синдром Марфана вызывается мутациями в гене FBN1 (15 хромосома), который кодирует гликопротеин фибрилин-1, являющийся компонентом внеклеточного матрикса. Белок фибрилин-1 имеет важное значение для правильного формирования внеклеточного матрикса, играет определенную роль при биогенезе и влияет на функционирование эластичных волокон. Кроме того, внеклеточный матрикс обеспечивает структурную целостность соединительной ткани, и играет роль резервуара для факторов роста (класс небольших природных пептидов и белков), главной целью которых является стимулирование роста клеток. Эластиновых волокон очень много во всем организме человека, но сконцентрированы они в основном в аорте, связках, в частности в цинновой связке (особая связка, с помощью которой хрусталик прикрепляется к цилиарному телу), именно поэтому эти части организма повреждаются при болезни Марфана больше всего. Для изучения механизма развития болезни Марфана была взята так называемая трансгенная мышь в организме которой в единственном экземпляре находился мутированный фибрилин-1 (мутации были аналогичны тем, которые происходят при изменении гена, кодирующего этот гликопротеин), который, как известно, является причиной развития синдрома Марфана. Этот вид мышей позволит изучить патогенез синдрома Марфана, ведь их черты болезни аналогичны человеческим. У мышей снижение нормального уровня фибрилина-1 приводит к развитию расстройств, связанных с болезнью Марфана.
Синдром Марфана существенно влияет на трансформирующий фактор роста бета (TGF-β). Ведь фибрилин-1, косвенно связывает неактивную форму TGF-β, будто поглощая ее, что в свою очередь приводит к снижению биологической активности этого фактора. Согласно простейшей схеме развития болезни Марфана, можно предположить, что снижение уровня фибрилина-1 приводит к увеличению количества TGF-β- из-за недостаточного его поглощения. И, хотя не доказано, что повышение уровня TGF-β приводит к возникновению патологий, связанных с синдромом Марфана, но, как известно, вследствие воспалительной реакции освобождения протеаз происходит постепенное ухудшение функциональности эластиновых волокон и других компонентов внеклеточного матрикса.
Достоверность данной гипотезы было подтверждена после открытия подобного синдрома с названием Синдром Лойса-Дитца (Syndrome Loeys-Dietz), который развивается при участии гена TGF-βR2 (3 хромосома), кодирующего рецептор к TGF-β. Через схожие клинические признаки эти два заболевания часто путают между собой.
Диагностика
Критерии, согласно которым диагностируют болезнь Марфана, были согласованы на международном уровне в 1996 году. Диагностика синдрома Марфана базируется на историях семей и на сочетании основных и второстепенных признаков болезни, которые в совокупности редко встречаются среди населения, но в единичном варианте могут проявляться в отдельной личности. Например - четыре нарушения со стороны опорно-двигательного аппарата сочетаются с патологиями других органов (органов системы зрения или сердечно-сосудистой системы) у одного человека - что может быть вероятным признаком синдрома Марфана.
Ниже перечисленные признаки могут быть вызваны болезнью Марфана, или могут возникнуть у людей, в которых любые нарушения отсутствуют:
• аневризма аорты или ее расширения;
• арахнодактилия;
• гастроэзофагеальная рефлюксная болезнь (ГЭРБ);
• двустворчатый аортальный клапан;
• кисты;
• кистозный медиальный некроз;
• дефекты перегородок сердца;
• дуальная ектазия;
• ранняя катаракта;
• ранняя глаукома;
• ранний остеоартрит;
• вывих хрусталика;
• эмфизема легких;
• колобома радужной оболочки;
• плоскостопие;
• рост - выше среднего;
• учащенное сердцебиение;
• грыжа;
• дисплазия суставов;
• кифоз (искривление верхнего отдела позвоночника.
• пролабирование клапанов сердца;
• неправильный прикус;
• микрогнатия (маленькая нижняя челюсть).
• пролапс митрального клапана;
• миопия (близорукость);
• обструктивные болезни легких;
• остеопения (пониженная плотность костной ткани);
• воронкообразное или килевидное искривление грудной клетки;
• пневмоторакс (коллапс легких);
• отслоения сетчатки;
• сколиоз;
• синдром апноэ;
• появление растяжек без причины (не от беременности, синдрома Кушинга или ожирения);
• нарушение роста зубов;
• узкое, худое лицо;
• артроз нижнечелюстного сустава.
Лечение
На сегодня лекарства от болезни Марфана не существует, однако за последние десятилетия продолжительность жизни с этим заболеванием значительно возросла, а клинические испытания, которые проводятся направленные на получение новых методов лечения и, на сегодня, довольно многообещающие. Синдром Марфана лечится по мере развития болезни, но особенно важным является профилактика заболевания, даже для маленьких детей, которая должна быть направлена на замедление развития аневризмы аорты.
Регулярные обзоры у кардиолога необходимые для контроля за состоянием здоровья сердечных клапанов и аорты. Цель лечения заключается в замедлении процессов прогрессирования аневризмы аорты и повреждение сердечных клапанов путем фармакологического устранения аритмий, уменьшение частоты сердечных сокращений (для чего могут быть использованы, например бета блокаторы) и снижения кровяного давления. Для его минимизации без снижения частоты сердечных сокращений, используют ингибиторы АПФ и антагонисты рецепторов ангиотензина II (также известные под названием блокаторы рецепторов ангионтензина или Сартана). Если диаметр расширение аорты становится существенным и развивается аневризма аорты, то это может стать причиной расслоения или разрыва аорты, или же приводит к недостаточности аортального или иного клапана. В таком случае необходимо проводить операцию (возможным, является трансплантация аортального клапана или проведения процедур для сохранения функциональности существующего клапана). Хотя хирургическая пересадка аорты, как и любая операция на сосудах достаточно серьезная задача, однако большинство из них можно считать успешными, если пациент добровольно и вовремя соглашается на сделку.
Операция в ситуации острого расслоения аорты или ее разрыва значительно опаснее и сложнее. Своевременным считается проведения операции на аорте, если диаметр ее корня составляет 50 мм, но каждый случай уникален и требует детальной оценки квалифицированного кардиолога.
На сегодняшний день все более распространенными становятся операции, с помощью которых осуществляется лечение клапанов. Также все более распространенными становятся методы лечения других сердечнососудистых заболеваний (нисходящей аневризмы грудной аорты и аневризмы других кровеносных сосудов). Именно поэтому, больные синдромом Марфана сегодня живут дольше, чем раньше.
Нарушение деятельности скелетной системы и системы зрения вследствие влияния болезни Марфана также серьезные, хотя и не опасны для жизни. Эти симптомы лечатся в обычном режиме, с помощью различных видов болеутоляющих средств или миорелаксантов. Очень важной в этом случае является работа физиотерапевта, который применяет TENS терапию (лечение током), ультразвук и различные способы улучшения деятельности скелетной системы, оказывает влияние на рост, длину рук и продолжительность жизни.
Физиотерапевт может также помочь улучшить функциональность опорно двигательной системы и уменьшить травматизм у лиц с болезнью Марфана. Для коррекции килевидного или воронкообразного искривления грудной клетки на западе применяются так называемые «Nuss»-процедуры. Так как очень часто болезнь Марфана приводит к патологиям позвоночника, то любое хирургическое вмешательство при осуществлении операции пациента с синдромом Марфана требует тщательного планирования и детальной визуальной диагностики различных отклонений современными методами.
Лечение спонтанного пневмоторакса, зависит от объема воздуха в плевральной полости и является уникальным для каждого больного. Если пневмоторакс незначительный, то его можно вылечить без активного вмешательства в течение одной-двух недель. Если же пневмоторакс постоянно повторяется, то больному может быть необходимо хирургическое вмешательство на грудной клетке. Для лечения пневмоторакса среднего размера нужно поставить грудной дренаж на несколько дней, конечно лечение должно осуществляться квалифицированным врачом. Если же пневмоторакс большой, то, вероятно, будет предоставлена быстрая медицинская помощь, в виде неотложной декомпрессии.
После исследования проведенного на мышах в лабораторных условиях было предположено, что антагонист рецепторов ангиотензина II лозартан, который, вероятно, блокирует активность TGF-бета, может замедлить или остановить развитие аневризмы аорты у больных с синдромом Марфана. Для сравнения эффекта действия лозартана и атенолола на аорту больных синдромом Марфана в 2007 году началось масштабное клиническое испытание под эгидой Национального института здоровья (США; деятельность которого координируется Университетом Джона Хопкинса
Также для помощи людям с синдромом Марфана и членам их семей действуют специализированные клиники и медицинские центры, проводятся генетические консультации.
Эпидемиология
Синдромом Марфана болеют как мужчины, так и женщины, при этом нет никаких этнических или географических особенностей данного заболевания. Согласно оценкам ученых один человек из 3000-5000 человек болеет синдромом Марфана. Через аутосомно-доминантный характер заболевания каждый из родителей может передать дефектные гены ребенку с вероятностью 50%. Большинство людей с синдромом Марфана имеют кого то в семье, кто уже поражен этим заболеванием, а 15-30% всех случаев связаны с новыми генетическими мутациями, которые возникают у одного ребенка на 20000 новорожденных. Синдром Марфана - это пример доминирующей негативной мутации и гаплонедостаточности. Вызванной переменной экспрессивностью, тогда как неполная пенетрантность на сегодня не является хорошо задокументированной.
Синдром Элерса - Данлоса

Синдром Элерса-Данлоса (несовершенный десмогенез, гиперэластическая кожа), наряду с несовершенным остеогенезом, синдромом Марфана и другими заболеваниями, относится к наследственным коллагенопатиям. Синдром Элерса-Данлоса неоднороден и включает в себя гетерогенную группу наследственных поражений соединительной ткани (соединительнотканных дисплазий), связанных с нарушением биосинтеза белка коллагена. Проявления синдрома Элерса-Данлоса носят системный характер и затрагивают опорно-двигательный аппарат, кожу, сердечно-сосудистую, зрительную, зубочелюстную и другие системы. Поэтому синдром Элерса-Данлоса представляет практический интерес не только для генетики, но и травматологии и ортопедии, дерматологии, кардиологии, офтальмологии, стоматологии.
Сложность верификации и наличие легких форм затрудняет получение точных сведений об истинной распространенности синдрома Элерса-Данлоса; частота диагностированных среднетяжелых случаев составляет 1:5 000 новорожденным, тяжелых форм - 1:100 000.
Причины синдрома Элерса-Данлоса
Различные варианты синдрома Элерса-Данлоса различаются по типу наследования, первичным молекулярным и биохимическим дефектам. Однако в основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена. На сегодняшний день молекулярные механизмы синдрома Элерса-Данлоса установлены не для всех форм заболевания.
Так, известно, что I тип синдрома характеризуется снижением активности фибробластов, усилением синтеза протеогликанов, отсутствием ферментов, отвечающих за нормальный биосинтез коллагена. Синдром Элерса-Данлоса IV типа связан с недостаточностью продукции коллагена III типа; при VI типе заболевания имеет место недостаточность фермента лизилгидроксилазы, участвующего в гидроксилировании лизина в молекулах проколлагена. VII тип обусловлен нарушением превращения проколлагена I типа в коллаген; X тип - патологией плазменного фибронектина, участвующего в организации межклеточного матрикса и т. п.
Патоморфологическая картина при различных типах синдрома Элерса-Данлоса характеризуется истончением дермы, нарушением ориентации и потерей компактности коллагеновых волокон, разрастанием эластических волокон, увеличением числа сосудов и расширением их просвета.
Классификация синдрома Элерса-Данлоса
Всего выделяют 10 типов синдрома Элерса-Данлоса, различающихся по генетическому дефекту, характеру наследования и клиническим проявлениям. Рассмотрим основные из них:
I тип синдрома Элерса-Данлоса (классический тяжелого течения) – наиболее частый вариант заболевания (43% случаев) с аутосомно-доминантным типом наследования. Ведущим симптомом является гиперэластичность кожи, растяжимость которой по сравнению с нормой увеличена в 2-2,5 раза. Характерна гипермобильность суставов, носящая генерализованный характер, деформации скелета, повышенная ранимость кожи, склонность кнаружным кровотечениям, образованию рубцов, плохому заживлению ран. У части больных выявляется наличие моллюскоподобных псевдоопухолей и варикозного расширения вен нижних конечностей. Беременность у женщин с I типом синдрома Элерса-Данлоса часто осложняется преждевременными родами.
II тип синдрома Элерса-Данлоса (классический мягкого течения) – характеризуется вышеописанными признаками, но выраженными в меньшей степени. Растяжимость кожи превосходит нормальную лишь на 30%; гипермобильность отмечается преимущественно в суставах стоп и кистей; кровоточивость и наклонность к рубцеванию незначительны.
III тип синдрома Элерса-Данлоса – имеет аутосомно-доминантное наследование, доброкачественное течение. Клинические проявления включают генерализованную повышенную подвижность суставов, скелетно-мышечные деформации. Остальные проявления (гиперэластичность и рубцевание кожи, геморрагии) минимальны.
IV тип синдрома Элерса-Данлоса – встречается редко, протекает тяжело; может наследоваться различными путями (доминантно или рецессивно). Гиперэластичность кожи незначительна, отмечается повышенная подвижность только суставов пальцев рук. Ведущим проявлением данного типа заболевания является геморрагический синдром: склонность к образованию экхимозов, спонтанных гематом (в т. ч. во внутренних органах), разрывам полых органов и сосудов (в т. ч. аорты). Сопровождается высокой летальностью.
V тип синдрома Элерса-Данлоса – имеет Х-сцепленное рецессивное наследование. Характеризуется повышенной растяжимостью кожи, умеренно выраженными гипермобильностью суставов, кровоточивостью и ранимостью кожи.
VI тип синдрома Элерса-Данлоса - наследуется по аутосомно-рецессивному типу. Кроме гиперэластичности кожи, наклонности к кровотечениям, повышенной подвижности суставов, имеются мышечная гипотония, тяжелыйкифосколиоз, косолапость. Характерной чертой синдрома Элерса-Данлоса VI типа является глазной синдром, проявляющийся близорукостью, кератоконусом, косоглазием, глаукомой, отслойкой сетчатки и т. д.
VII тип синдрома Элерса-Данлоса (артроклазия) - наследуется как аутосомно-доминантно, так и аутосомно-рецессивно. Клиническую картину определяет низкий рост пациентов и гиперподвижность суставов, приводящая к частым привычным вывихам.
VIII тип синдрома Элерса-Данлоса – преимущественно наследуется аутосомно-доминантно. Ведущую роль в клинике играет хрупкость кожи, выраженный периодонтит, приводящий к ранней потере зубов.
X тип синдрома Элерса-Данлоса – характеризуется аутосомно-рецессивным наследованием; умеренной гиперэластичностью кожи и гипермобильностью суставов, стриями (полосовидной атрофией кожи), нарушением агрегации тромбоцитов.
XI тип синдрома Элерса-Данлоса – имеет аутосомно-доминантный тип наследования. У больных отмечаются рецидивирующие вывихи плечевых суставов, вывихи надколенника, встречается врожденный вывих бедра.
IX тип (Х-спепленный вариант вялой кожи) в настоящее время исключен из классификации синдрома Элерса-Данлоса. В современном варианте классификации синдрома Элерса-Данлоса рассматривается 7 основных типов заболевания:
· классический (типы I и II)
· гипермобильный (тип III)
· сосудистый (тип IV)
· кифосколиоз (тип VI)
· артроклазия (тип VIIB)
· дермоспараксис (тип VIIC)
· недостаток тенасцина-X
Симптомы синдрома Элерса-Данлоса
Ввиду того, что подробная характеристика различных типов синдрома Элерса-Данлоса дана выше, в настоящем разделе обобщим основные проявления заболевания. Поскольку соединительная ткань присутствует практически во всех органах, проявления синдрома Элерса-Данлоса носят системный, генерализованный характер.
Ведущим в клинической картине является кожный синдром: гиперэластичность кожи, которая легко собирается в складку и оттягивается. На ощупь кожа бархатистая, нежная, слабо фиксированная с подлежащими тканями, морщинистая на ладонных и подошвенных поверхностях. Гиперэластичность кожи при синдроме Элерса-Данлоса отмечается с рождения или дошкольного возраста, с годами имеет тенденцию к снижению.
Кроме, гиперрастяжимости, характерна повышенная ранимость, хрупкость кожи, обнаруживающаяся в возрасте старше 2-3-х лет. Минимальная травматизация приводит к образованию длительно не заживающих ран, на месте которых спустя время формируются атрофичные или келоидные рубцы, псевдоопухоли.
Суставные проявления синдрома Элерса-Данлоса представлены гипермобильностью (разболтанностью) суставов, которая может носить локальный (например, переразгибание межфаланговых суставов) или генерализованный характер. Суставной синдром проявляется с началом ходьбы ребенка, что приводит к повторным подвывихам и вывихам. С возрастом гипермобильность суставов обычно уменьшается.
Со стороны сердечно-сосудистой системы у детей с синдромом Элерса-Данлоса нередко выявляютсяврожденные пороки сердца, пролапс митрального клапана, аневризмы сосудов головного мозга, варикоз. Отмечается склонность к кровотечениям - экхимозам, гематомам различной локализации, носовым, десневым,маточным, желудочно-кишечным кровотечениям.
Глазные проявления синдрома Элерса-Данлоса могут включать гиперэластичность кожи век, миопию, птоз, косоглазие, разрывы роговицы и глазного яблока при минимальных механических повреждениях, спонтанную отслойку сетчатки.
Изменения скелета при синдроме Элерса-Данлоса характеризуются воронкообразной или килевиднойдеформацией грудной клетки, сколиозом, кифозом, косолапостью, неправильным прикусом, частичной адентией. Висцеральные нарушения представлены птозом внутренних органов, пупочными, паховыми, диафрагмальными грыжами, рецидивирующим спонтанным пневмотораксом, дивертикулезом кишечника и др. Умственное развитие детей с синдромом Элерса-Данлоса обычно соответствует возрасту.
Диагностика синдрома Элерса-Данлоса
Диагностика синдром Элерса-Данлоса проводится медицинским генетиком на основании генеалогических данных, анамнеза, клинического анализа, молекулярно-генетических исследований. Предварительно синдром Элерса-Данлоса может быть заподозрен при наличии больших диагностических критериев (гипермобильности суставов, гиперэластичности кожи, склонности к кровотечениям) и дополнительных малых (хрупкости кожи, патологии сердца, сосудов, глаз и т. д.).
Некоторые формы заболевания требуют проведения биопсии кожи для гистологического, гистохимического, электронно-микроскопического исследования.
Наличие в семье больных синдромом Элерса-Данлоса является показанием к медико-генетическому консультировании и проведению инвазивной пренатальной диагностики.
Больные с различными типами синдром Элерса-Данлоса могут нуждаться в наблюдении и обследованиидетским травматологом-ортопедом, детским кардиологом, детским офтальмологом, детским стоматологом,сосудистым хирургом.
Лечение синдрома Элерса-Данлоса
Эффективная специфическая терапия синдрома Элерса-Данлоса не разработана. Детям требуется создание щадящего режима, исключающего излишнюю травматизацию суставов и кожи; ограничение физических нагрузок; соблюдение белковой диеты с включением в рацион костных бульонов, заливных блюд, студня. Обязательны регулярные курсы массажа, лечебной физкультуры, физиотерапии (магнитотерапии, электрофореза,лазеропунктуры).
Медикаментозная терапия синдрома Элерса-Данлоса включает применение аминокислот (карнитина, нутраминоса), витаминов (С, Е, D, группы В), хондроитина сульфата, глюкозамина, минеральных комплексов (препаратов кальция и магния), метаболических препаратов (рибоксин, АТФ, коэнзим Q10) повторными курсами до1-1,5 мес. 2-3 раза в год.
При синдроме Элерса-Данлоса может быть показано хирургическое лечение: реконструкция грудной стенки, удаление псевдоопухолей, коррекция ВПС и пр.
Прогноз синдрома Элерса-Данлоса
На качество и продолжительность жизни больных синдромом Элерса-Данлоса влияет тип заболевания. Наиболее серьезный прогноз имеет IV тип синдрома Элерса-Данлоса – летальный исход может наступить вследствие разрывов сосудов, внутренних органов и кровотечений. Наличие синдрома I типа существенно ограничивает качество жизни. Относительно благоприятно протекание II—III типов болезни.
В целом, наличие синдрома Элерса-Данлоса сопряжено со множеством социальных трудностей, ограничивает полноценную физическую активность и выбор профессии.
Ахондроплазия
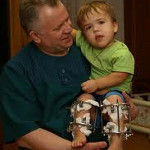
Ахондроплазия (врожденная хондродистрофия, болезнь Парро-Мари, диафизарная аплазия) – врожденное заболевание, при котором нарушается процесс роста костей. Поражаются кости скелета и основания черепа. Причиной развития ахондроплазии является мутация гена FGFR3. В 20% случаев ахондроплазия передается по наследству, в 80% развивается в результате впервые возникшей мутации. Часть плодов с такой патологией гибнет внутриутробно. При рождении нарушения заметны с первых дней жизни: головка увеличена, руки и ноги укорочены. В последующем наблюдается выраженное отставание в росте конечностей при нормальном размере туловища, возникают вальгусные и варусные деформации конечностей, а также деформации позвоночника. Из-за скелетной патологии могут развиваться вторичные нарушения со стороны внутренних органов. Лечение ахондроплазии симптоматическое, направлено на предотвращение и устранение грубых деформаций.
Ахондроплазия
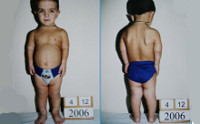
Ахондроплазия – генетическое заболевание, при котором наблюдается укорочение конечностей в сочетании с нормальной длиной туловища. Характерными особенностями являются низкий рост (130 и менее см.), изогнутый вперед позвоночник, седловидный нос и относительно большая голова с выступающими лобными буграми. Ахондроплазия возникает у одного из 10 тысяч новорожденных, женщины страдают чаще мужчин. Способов полностью излечить ахондроплазию, восстановив рост и пропорции тела, в настоящее время не существует. Лечение направлено на минимизацию негативных последствий болезни.
Причины развития ахондроплазии
В основе ахондроплазии лежит нарушение развития костей вследствие генетически обусловленной дистрофии эпифизарных хрящей. Из-за хаотичного расположения клеток ростковой зоны происходит нарушение нормального процесса окостенения. В результате рост костей замедляется. При этом поражаются только кости, растущие по энхондральному типу: трубчатые кости, кости основания черепа и т. д. Кости свода черепа, растущие из соединительной ткани, достигают положенного размера, что приводит к несоответствию пропорций между головой и телом, а также становится причиной характерного изменения формы черепа.
Симптомы ахондроплазии
Нарушение анатомических пропорций заметно уже при рождении: ребенок с ахондроплазией имеет от