Пояснительная записка к магистерской диссертации
(выпускной квалификационной работе)
на тему:
Разработка методов и средств статистического моделирования биотехнологических процессов
Студент-дипломник: ________________________ / _______________ /
Руководитель проекта: ________________________ / _______________ /
Рецензент: ________________________ / _______________ /
Рецензент: ________________________ / _______________ /
Заведующий кафедрой №12: ________________________ / _______________ /
Москва 2013
АННОТАЦИЯ
Данная работа направлена на решение задач биоинформатики в области изучения ДНК-полиморфизма. Основной целью работы является разработка эффективной системы эвристического поиска видоспецифичных молекулярно-генетических маркеров, позволяющих эффективно выявлять ДНК-полиморфизм у различных организмов, геном которых полностью секвенирован. Работа состоит из трех основных глав, а также включает введение, заключение и приложение с листингами разработанных программ.
Во введении дается краткое описание проблемы, на решение которой направлена работа. Оценивается актуальность исследований в данной области. Формулируются основные цели и задачи исследования.
В главе 1 дается описание типов молекулярно-генетических маркеров и требований, предъявляемых к ним. Приводятся обобщенные данные по методам определения ДНК-полиморфизма с помощью полимеразной цепной реакции. Дается описание основным форматам представления биологических последовательностей, таким как FASTA, FASTQ и GenBank. Описываются наиболее распространенные программные средства анализа нуклеотидных и аминокислотных последовательностей.
В главе 2 обсуждаются принципы разрабатываемого метода поиска молекулярно-генетических маркеров. Дается обоснование условиям поиска и критериям отбора результатов. Вводится понятие критерия оценки приоритетности использования праймеров для изучения ДНК-полиморфизма – TVmax. Приводится описание реализации на языке Си алгоритмов поиска праймеров на основе полногеномных последовательностей по заданному префиксу, виртуальной ПЦР и сортировки полученных праймеров в порядке снижения их приоритетности для использования в ПЦР. Дается оценка производительности алгоритма виртуальной ПЦР по результатам профилирования. Описывается реализация на Си следующих алгоритмов поиска подстроки в строке: примитивного алгоритма, алгоритма Кнута-Морриса-Пратта, алгоритма Бойера-Мура, турбо-алгоритма Бойера-Мура, алгоритма Чжу-Такаоки, алгоритма GRASPM, алгоритма быстрого поиска, алгоритмов сдвига-или и Карпа-Рабина, а также ассемблерные реализации примитивного алгоритма и алгоритма Кнута-Морриса-Пратта. Приводятся результаты сравнения быстродействия этих алгоритмов, с библиотечной функцией strstr. Описывается принцип распараллеливания циклического участка алгоритма виртуальной ПЦР на основе технологии Open Multi-Processing.
В главе 3 описываются условия проведения тестирования программ по разработанным алгоритмам на примере поиска праймеров для выявления полиморфизма у растения Arabidopsis thaliana (L.) Дается характеристика объекта исследования. Описываются условия поиска и полученные результаты.
В заключении дается оценка: работоспособности алгоритмов, результатам экспериментов и применимости данного подхода для подбора молекулярно-генетических маркеров. Делаются выводы по данной работе.
В приложении представлен полный текст исходного кода программ на языке Си по разработанным алгоритмам: первичного поиска праймеров, поиска теоретических ПЦР-фрагментов по найденным праймерам и отбора оптимальных праймеров. Также представлены листинги всех алгоритмов поиска подстроки, используемых в работе, и алгоритма сортировки с использованием 2-3 дерева.
ОГЛАВЛЕНИЕ
1. Аналитический обзор литературы.. 8
1.1. Молекулярно-генетические маркеры.. 8
1.1.1. Требования, предъявляемые к молекулярным маркерам.. 8
1.1.2. Анализ полиморфизма с помощью ПЦР.. 9
1.2. Стандартные форматы представления биологических последовательностей. 19
1.2.1. Формат FASTA.. 19
1.2.2. Формат FASTQ.. 22
1.2.3. Формат GenBank. 23
1.3. Биоинформационное программное обеспечение. 23
1.3.1. Программное обеспечение Vector NTI. 24
1.3.2. Программное обеспечение UGENE.. 26
1.3.3. Программное обеспечение Primer Express. 28
1.3.4. Программное обеспечение OLIGO Primer Analysis Software. 28
1.3.5. Программное обеспечение Visual Cloning 2000. 28
1.3.6. Программное обеспечение DnaSP.. 31
Выводы к первой главе. 31
2. Разработка алгоритмов поиска праймеров для идентификации полиморфных фрагментов 33
2.1. Обоснование выбора условий поиска и критериев сортировки праймеров. 33
2.2. Приведение исходных данных к виду удобному для проведения поиска. 37
2.2.1. Преобразование исходного формата FASTA.. 38
2.2.2. Корректировка возможных ошибок и недопустимых значений входных данных 39
2.3. Способы представления промежуточных данных и результатов эксперимента. 40
2.4. Алгоритм первичного поиска праймеров. 41
2.5. Сортировка массива праймеров с использованием 2-3 дерева. 42
2.6. Алгоритм поиска теоретических ПЦР-фрагментов по найденным праймерам.. 45
2.6.1. Выявление участков кода, требующих оптимизации. 48
2.6.2. Обзор алгоритмов точного поиска подстроки в строке. 49
2.6.2.1. Примитивный алгоритм.. 50
2.6.2.2. Алгоритм Кнута-Морриса-Пратта. 51
2.6.2.3. Реализация примитивного алгоритма поиска на ассемблере. 53
2.6.2.4. Реализация алгоритма Кнута-Морриса-Пратта на ассемблере. 55
2.6.2.5. Алгоритм Бойера-Мура. 58
2.6.2.6. Турбо-алгоритм Бойера-Мура. 63
2.6.2.7. Алгоритм Чжу-Такаоки. 65
2.6.2.8. Алгоритм GRASPM... 67
2.6.2.9. Алгоритм быстрого поиска. 68
2.6.2.10. Алгоритм сдвига-или. 69
2.6.2.11. Алгоритм Карпа-Рабина. 71
2.6.3. Сравнение быстродействия алгоритмов при заданных условиях поиска и структуре программы.. 73
2.7. Автоматическое распараллеливание цикла for. 74
2.8. Алгоритм отбора оптимальных праймеров. 78
Выводы ко второй главе. 79
3. Отработка программ по разработанным алгоритмам на примере поиска праймеров для растения Arabidopsis thaliana (L.) 81
3.1. Характеристика объекта исследования. 81
3.2. Входные данные и условия поиска. 81
3.3. Анализ результатов поиска. 82
Выводы к третьей главе. 85
ЗАКЛЮЧЕНИЕ.. 86
СПИСОК ЛИТЕРАТУРЫ... 88
ПРИЛОЖЕНИЕ.. 94
Исходный код программы преобразования формата FASTA на Си. 94
Исходный код программы первичного поиска праймеров на Си. 95
Исходный код программы сортировки с использованием 2-3 дерева на Си. 97
Исходный код программы поиска теоретических ПЦР-фрагментов по найденным праймерам на Си 105
Исходный код программы поиска теоретических ПЦР-фрагментов по найденным праймерам с распараллеленным циклом на Си. 118
Исходный код программы отбора оптимальных праймеров на Си. 131
Исходный код ассемблерной реализации примитивного алгоритма. 133
Исходный код ассемблерной реализации алгоритма Кнута-Морриса-Пратта. 134
ВВЕДЕНИЕ
Разнообразие организмов обусловлено изменчивостью последовательностей ДНК и влиянием факторов среды. Генетическая изменчивость значительна, и каждый организм данного вида, за исключением клонов, несет уникальные последовательности ДНК. ДНК-варианты являются следствием мутаций, происходящих вследствие замены одного нуклеотида (однонуклеотидный полиморфизм – single nucleotide polymorphisms, SNP), вставок или потерь фрагментов ДНК разной длины (от одного до нескольких тысяч пар нуклеотидов), а также дупликаций или инверсий фрагментов ДНК.
Выявление этого разнообразия позволяет решать широкий спектр задач, таких как:
• Оценка генеалогических связей;
• Идентификация сортов растений и пород животных;
• Поиска маркеров, ассоциированных с желательными признаками, которые подвергаются воздействию факторов искусственного и естественного отбора;
• Оценки изменчивости как внутри популяции, так и между различными популяциями;
• Выявления воздействия на геном неблагоприятных факторов окружающей среды.
Для выявления такого генетического разнообразия используются различные типы молекулярно-генетических маркеров. На данный момент наибольшее распространение получили методы определения ДНК-полиморфизма с помощью полимеразной цепной реакции. На этом принципе основано несколько методов, отличающихся по способу подбора специфичных нуклеотидных последовательностей – праймеров, необходимых для протекания реакции. Общим недостатком таких методов является отсутствие четкой стратегии подбора праймеров, в результате чего для получения качественного и воспроизводимого фингерпринта необходима экспериментальная проверка сотен праймеров и их комбинаций и отбор из них наиболее удачных. Таким образом, разработка эффективной системы подбора ПЦР-праймеров для выявления полиморфизма является актуальной задачей.
Целью настоящей работы являлось разработка эффективной системы эвристического поиска видоспецифичных молекулярно-генетических маркеров, позволяющих эффективно выявлять ДНК-полиморфизм у различных организмов, геном которых полностью секвенирован.
Для достижения поставленной цели необходимо было решить следующие задачи:
· Определить входные данные и условия поиска;
· Разработать алгоритмы, позволяющие осуществлять подбор праймеров для ПЦР;
· Разработать критерии оценки отобранных праймеров, по пригодности для использования в качестве молекулярно-генетических маркеров;
· Провести тестирование разработанных алгоритмов на реальном объекте исследований.
Требования, предъявляемые к молекулярным маркерам
Используемые молекулярно-генетические маркеры должны обладать определенными свойствами и отвечать ряду требований [5, 14, 26, 51, 52]:
· доступность фенотипических проявлений аллельных вариантов для идентификации у разных особей;
· отличимость аллельных замещений в одном локусе от таковых в других локусах;
· доступность существенной части аллельных замещений в каждом изучаемом локусе для идентификации;
· изучаемые локусы должны представлять случайную выборку генов в отношении их физиологических эффектов и степени изменчивости;
· равномерность распределения по локализации в геноме;
· легкая выявляемость, воспроизводимость и дешевизна;
· возможность автоматизации выявления;
· относительная нейтральность.
Очевидно, что не существует такого стандартного набора маркеров, который отвечал бы всем этим требованиям.
Формат FASTA
FASTA — один из наиболее популярных тестовых форматов для представления нуклеотидных и аминокислотных последовательностей, в котором нуклеотиды или аминокислоты кодируются при помощи одного символа. Данный формат был впервые использован в программном обеспечении FASTA [53], но позже стал общепринятым стандартом в области биоинформатики.
Файл в формате FASTA [16] представляет собой простой текстовый файл, первая строка которого должна начинаться с символа ״>״ или ״;״. Эта строка содержит идентификатор (имя) последовательности и некоторую вспомогательную информацию, необходимую для краткого описания последовательности. Другие строки, начинающиеся с символа ״;״ являются комментариями и игнорируются программным обеспечением. Впоследствии от использования символа ״;״ в начале файла и от дополнительных комментариев, начинающихся с этого же символа, отказались и стали использовать только символ ״>״ и одну единственную первую строку для идентификации последовательности и комментариев. При этом между знаком ״>״ и первым символом в имени последовательности не должно быть никаких вспомогательных символов или знаков.
Таблица 1.1.
Обозначение нуклеотидов в формате FASTA
| Код нуклеиновой кислоты
| Значение
| Обозначение
|
| A
| A
| аденин
|
| C
| C
| цитозин
|
| G
| G
| гуанин
|
| T
| T
| тимин
|
| U
| U
| урацил
|
| R
| A или G
| пуриновые основания
|
| Y
| С или T или U
| пиримидиновые основания
|
| K
| G или T или U
| -
|
| M
| A или C
| -
|
| S
| C или G
| Сильное взаимодействие
|
| W
| A или T или U
| Слабое взаимодействие
|
| B
| не A
| любое, кроме A
|
| D
| не C
| любое, кроме C
|
| H
| не G
| любое, кроме G
|
| V
| не T и не U
| Любое, кроме Т и U
|
| N
| A или С или G или T или U
| любой
|
| X
| замаскированный
| -
|
| -
| неизвестная последовательность произвольной длины
| -
|
После строки с заголовком и комментариями начинается описание последовательности. Оно представлено в виде одной или нескольких строк с буквенным кодом. Рекомендуется, чтобы каждая строка состояла не более чем из 80 символов, хотя возможно использование и более длинных строк (до 132 символов). При кодировании последовательности нуклеотидов, буквы A, C, G, T и U кодируют, соответственно, аденин, цитозин, гуанин, тимин и урацил. Также некоторые буквы (табл. 1.1.) кодируют позиции, в которых находится один нуклеотид из некоторого множества (это используется, если точно неизвестно, какой именно нуклеотид там находится). Символы, кодирующие аминокислоты, представлены в таблице 1.2. Символ ״-״ (дефис) кодирует неизвестную последовательность произвольной длины.
Таблица 1.2.
Обозначение аминокислот и специальных триплетов
| Код
| Описание
|
| A
| Аланин
|
| B
| Аспарагиновая кислота или Аспарагин
|
| C
| Цистеин
|
| D
| Аспарагиновая кислота
|
| E
| Глутаминовая кислота
|
| F
| Фенилаланин
|
| G
| Глицин
|
| H
| Гистидин
|
| I
| Изолейцин
|
| K
| Лизин
|
| L
| Лейцин
|
| M
| Метионин
|
| N
| Аспарагин
|
| O
| Пирролизин
|
| P
| Пролин
|
| Q
| Глутамин
|
| R
| Аргинин
|
| S
| Серин
|
| T
| Треонин
|
| U
| Селеноцистеин
|
| V
| Валин
|
| W
| Триптофан
|
| Y
| Тирозин
|
| Z
| Глутаминовая кислота или Глутамин
|
| X
| любая
|
| *
| остановка трансляции
|
| -
| неизвестная последовательность произвольной длины
|
В строках последовательности допустимо использование пробелов, табуляции и символа ״*״, которые, как правило, игнорируются программным обеспечением. Пробелы и табуляция служат для выравнивания последовательностей, а символ ״*״ является признаком конца последовательности. Позже этот символ был заменен на пустую строку. Если в файле встречается несколько символов ״>״, то каждый из них будет указывать на начало новой последовательности.
Формат FASTQ
FASTQ - формат представления биологической последовательности (обычно, последовательности нуклеотидов) совместно с данными об их качестве [33].
Этот формат используется для представления данных секвенирования, так как позволяет представить как саму последовательность, так и вероятность, что каждый из элементов последовательности указан правильно. Для этого, кроме символов, кодирующих элементы последовательности, используется символы, кодирующие уровень качества.
Уровень качества — целое число в некотором диапазоне. Известно два различных способа выражать уровень качества через вероятность ошибки. Чаще всего используется следующая формула (1.1):
 (1.1)
(1.1)
Здесь Q - уровень качества, а p - вероятность, что этот элемент последовательности ошибочный.
Реже применяется второй способ (1.2):
 (1.2)
(1.2)
При больших значениях Q (и, соответственно, при малых p) эти способы дают практически идентичные результаты, но при малых значениях Q (Q < 13) и больших значениях p (p > 0,05) их значения заметно различаются.
Сам же файл содержит четыре строки для каждой последовательности. Первая строка начинается с символа ״@״, после которого идёт идентификатор (имя) и описание последовательности, как и в формате FASTA. Следующая строка содержит последовательность символов, кодирующих саму последовательность, также аналогично формату FASTA. Третья строка, начинающаяся с символа ״+״, после которого может идти описание последовательности (третья строка будет отличаться от первой только тем, что первый символ заменён на ״+״). Зачастую эта строка содержит лишь символ ״+״ без какого-либо описания.
Последняя строка кодирует уровни качества. Её длина равна длине второй строки, а каждый символ кодирует информацию о качестве (вероятности) элемента последовательности, закодированного соответствующим символом во второй строке. Сами же уровни кодируются таким образом: ASCII-код символа равен уровню качества плюс некоторая константа. Константа обычно имеет значение 33 или 64. В любом случае, код символа не должен превышать 127.
Формат GenBank
Формат GenBank позволяет представлять больше дополнительной информации о последовательностях [17]. Файл в формате GenBank состоит из нескольких записей, каждая из которых может занимать несколько строк. Все строки в записи, кроме первой, начинаются с пробела, это позволяет легко находить границы записей.
Каждая запись начинается с заголовка (идентификатора), за которым идёт значение (через один или несколько пробелов). Некоторые записи могут содержать подзаписи. Они форматируются аналогично записям. После этого в начало каждой строки записывается несколько пробелов. Таким образом, значения подзаписей выровнены правее значений записей, а те выровнены правее имён записей.
Имена записей имеют предопределённое значение. Например, запись с заголовком DESCRIPTION хранит описание последовательности, запись с заголовком SOURCE идентифицирует объект, из которого взята последовательность, а запись с заголовком REFERENCE (их может быть несколько) используется для описания ссылок на публикации.
Запись с заголовком ORIGIN содержит саму последовательность. Каждая строка, кроме последней, содержит 60 элементов, разбитых пробелами на группы по 10. Для представления элементов используются строчные буквы. В начало каждой строки добавляется текущая позиция, начиная с единицы, и пробел.
Помимо трех основных форматов FASTA, FASTQ и GenBank, в биоинформатике существует еще множество менее распространенных текстовых форматов представления данных биологических последовательностей, таких как: EMBL, GCG, GCG-RSF, IG и др. [15].
Выводы к первой главе
Описанные выше программные продукты представляют собой далеко не полный список ПО, позволяющего выполнять всесторонний анализ нуклеотидных и аминокислотный последовательностей, а являются лишь наиболее широко известными. Однако, не смотря на большое разнообразие программ и алгоритмов по анализу последовательностей, до сих пор не существует методов позволяющих на основе эвристических подходов подбирать молекулярно-генетические маркеры для выявления полиморфизма ДНК при помощи ПЦР. В настоящее время подбор оптимальных МГМ для ПЦР (праймеров, дающих при постановке ПЦР максимальное количество четко различимых полос) осуществляется экспериментальным перебором множества уже известных праймеров (ранее использованных на других объектах), или множества праймеров подобранных на основе идентифицированных последовательностей, или же, наконец, множества случайных коротких праймеров. При этом материальные и трудовые затраты на поиск эффективных МГМ зачастую превосходят затраты на дальнейшее изучение полиморфизма из-за огромного количества проверяемых праймеров. При этом, если ограничить количество проверяемых праймеров, то среди них можно вообще не обнаружить хороших МГМ. К тому же эффективность применения МГМ зачастую видоспецифична, и простой перенос, пусть даже хороших маркеров, с одного объекта на другой часто не приводит к положительным результатом.
Поэтому создание эффективной системы эвристического подбора видоспецифичных молекулярно-генетических маркеров является актуальной задачей, решение которой невозможно без применения современных вычислительных средств.
Примитивный алгоритм
Примитивный алгоритм или алгоритм грубой силы, заключается в проверке всех позиций текста с 0 по n - m на предмет совпадения с началом образца. Если совпадает - смотрим следующую букву и т.д., если же не совпадает, то смещаемся на один символ в строке y и начинаем сравнение заново. Этот алгоритм не нуждается в предварительной обработке и дополнительных затратах памяти:
char* hospool(char *y, char *x, int m){
char* yy;
int i, j;
yy=y;
i=0;
while (y[i]!= '\0'){
for (j=0; j < m; ++j){
if (y[i+j]!= x[j])
break;
}
if (j==m){
yy=y+i;
return (yy);
}
i++;
}
return NULL;
}
Данный алгоритм имеет среднее время поиска равное 2*n, и худшее время поиска - O(n * m)
Алгоритм Бойера-Мура
Данный алгоритм разработанный двумя учеными Робертом Бойером и Джем Муром в 1977 году, считается наиболее быстрым среди алгоритмов общего назначения, предназначенных для поиска подстроки в строке [29]. Преимущество этого алгоритма в том, что ценой некоторого количества предварительных вычислений над шаблоном (но не над строкой, в которой ведётся поиск) шаблон сравнивается с исходным текстом не во всех позициях - часть проверок пропускаются как заведомо не дающие результата.
Данный алгоритм совершает 3n сравнений в худшем случае при поиске первого совпадения с непериодничным образцом и O(n*m) при поиске всех вхождений. В алгоритме Бойера-Мура образец движется вдоль строки текста слева направо, однако фактические сравнения символов выполняются справа налево. В случае несовпадения (или, наоборот, полного попадания) используются две заранее вычисляемые функции: функция плохого символа и функция хорошего суффикса.
Первыми, таким образом, сравниваются символы x [ m-1 ] и y [ m-1 ]. Если они не совпадают и y [ m-1 ] не встречается нигде в подстроке x, мы можем спокойно сдвинуть образец на m символов вправо, так как можем быть уверены, что ни одна из начинающихся с одной из первых m позиций подстрок не совпадает с образцом. Следующими сравниваются x [ m-1 ] и y [ 2*m-1 ].
Пусть отвергающими оказались символы x [ m-1 ] и y[ i ]. Как уже говорилось, если y [ i ] не содержится нигде в образце, шаблон можно сдвинуть на m позиций вправо. С другой стороны, если y [ i ] присутствует в образце, и x [ m-s-1 ] – его самое правое вхождение, то образец можно сдвинуть лишь на s позиций вправо, совмещая x[ m-s-1 ] с y [ i ]. Проверку можно продолжить сравнением x [ m-1 ] с y [ i+s ], то есть текстовый индекс мы увеличиваем на s.
Если x [ m-1 ] и y [ i ] совпадают, нужно сравнивать предшествующие им символы в образце и тексте, пока вся подстрока текста не будет сопоставлена с образцом или пока не встретится несовпадение. Предположим, что несовпадение произошло между символом образца x [ j ] = b и символом текста y [ i+j ] = a на позиции i, то мы знаем, что y [ i+j+1, i+m-1 ] = x [ j+1, m-1 ] = u и y [ i+j ] ≠ x [ j ].
Теперь, если самым правым вхождением y [ i+j ] в образец снова является, скажем, x [ m-s-1 ], образец можно сдвинуть на j-m+s позиций вправо, так что x [ m-s-1 ] окажется на одной позиции с y [ i+j ]. Процедуру можно продолжить сравнением x [ m-1 ] с соответствующим символом, в данном случае с y [ i+j+s ]. Приращение индекса текста с позиции несовпадения до следующей пробной позиции равно, таким образом, [ i+j+s ] - [ i+j ] = s.
Если x [ m-s-1 ] находится правее x [ j ], то значение j-m+s отрицательно, так что совмещение x [ m-s-1 ] с y [ i+j-s ] ничего не даст, поскольку повлечет за собой шаг назад. В этих обстоятельствах образец лучше сдвинуть на одно место вправо и сравнивать x [ m-1 ] с y [ i+1 ]. Для этого нужно увеличить индекс текста на [ i+1 ] - [ i+j-m ] = j-m+1.
Таким образом, функция плохого символа выравнивает символ текста y [ i + j ] по его самому правому появлению в x [ 0... m-s-1 ]. Если его там нет - сдвигаем на всю длину образца: левый конец теперь - y [ i + j + m ].
Приращения s, используемые в эвристике вхождения, вычисляются заранее и хранятся в таблице bmBc. В процессе поиска таблица индексируется отвергающими символами текста, поэтому требуется таблица размером с используемый алфавит символов, то есть SIGMA. Вход для символа w равен m-j, где x[ j ] – самое правое вхождение w в x, и равен m-1, если w не содержится в x, то есть:
bmBc[
w ] = min {
s:
s =
m или (
0 ≤ s < m и
x [
m-s-1 ] =
w)}
Если, как в описанном выше случае, несовпадение встречено уже после того, как найдено частичное совпадение, иногда можно продвинуться дальше, чем позволяет эвристика вхождения. Если это так, лучше взять больший сдвиг, определяемый эвристикой сопоставления. При этом используется таблица, похожая на используемую в алгоритме Кнута-Морриса-Пратта. Идея состоит в том, что в результате сдвига символы образца должны быть равны всем тем символам текста, с которыми они совпадали перед этим, а на месте отвергающего символа должен оказаться другой.
Как уже говорилось, особый интерес представляет ситуация, когда часть образца содержится в текущей части текста, то есть x [ j+1, m-1 ] = y [ i+j+1, i+m-1 ] и x [ j ] ≠ y [ i+j ]. Если суффикс x [ j+1, m-1 ] входит также в x как подстрока x [ j+1-t, m-t-1 ], причем x [ j-t ] ≠ x [ j ], и это самое правое из подобных вхождений, то образец можно сдвинуть на t мест вправо. При этом мы совмещаем x [ j+1-t, m-t-1 ] с y [ i+j+1, i+m-1 ] и процесс поиска можно возобновить, сравнивая x[ m-1 ] с y[ i+t ]. Таблица bmGs, содержащая информацию о сдвиге t, вычисляется заранее на основании образца и индексируется той позицией образца, в которой встретилось несовпадение. Значение bmGs[ j ] равняется сдвигу t образца плюс дополнительный сдвиг, требуемый для того, чтобы возобновить сравнение от правого края образца. Значение i+m-1 – это минимум, такой что, если x [ j ] ≠ y [ i+j ], то x [ j+1-t, m-t-1 ] совпадет с y [ i+j+1, i+m-1 ]. Итак, входы таблицы bmGs определяются следующим выражением:
bmGs [ j ] = min{ t + m - j: t ≥ 1 и (t ≥ j или x [ j-t ] ≠ x [ j ]) и ((t ≥ k или x [ k-t ] = x [ k ]) для j < k ≤ m)}
Таким образом, значение bmGs [ j ] равно требуемому приращению для индекса текста с текущей позиции y[ i+j ] до позиции следующего сравнения y[ i+t ], то есть [ i+t ] - [ i+j ] = t-j.
Следовательно функция хорошего суффикса состоит в выравнивании сегмента y [ i+j+1, i+m-1 ] = x [ j+1, m-1 ] = u по его самому правому появлению в x, так чтобы ему предшествовал символ, несовпадающий с x [ j ] (рис. 2.8.). Если такого сегмента нет, то сдвиг выравнивает наибольший суффикс v отрезка y [ i+j+1,i+m-1 ] по совпадающему префиксу x.
Из определения bmGs [ j ] можно углядеть, что bmGs [ j ] ≥ [ m-j ]. Так, в указанном выше случае, где индекс текста следует увеличить на m-j , вместо соответствующего значения bmGs, так как последнее вызывает сдвиг образца назад. Таким образом, в случае несовпадения следует использовать максимальное из двух значений, взятых из описанных выше таблиц.

Рисунок 2.8. Осуществление сдвига в алгоритме Бойера-Мура.
Исходный код реализации алгоритма Бойера-Мура имеет следующий вид:
/*Построение таблицы для сдвига по функции плохого символа*/
void preBmBc(unsigned char *x, int m, int bmBc[]) {
int i;
for (i = 0; i < SIGMA; ++i)
bmBc[i] = m;
for (i = 0; i < m - 1; ++i)
bmBc[x[i]] = m - i - 1;
}
void suffixes(unsigned char *x, int m, int *suff) {
int f, g, i;
suff[m - 1] = m;
g = m - 1;
for (i = m - 2; i >= 0; --i) {
if (i > g && suff[i + m - 1 - f] < i - g)
suff[i] = suff[i + m - 1 - f];
else {
if (i < g)
g = i;
f = i;
while (g >= 0 && x[g] == x[g + m - 1 - f])
--g;
suff[i] = f - g;
}
}
}
/*Построение таблицы для сдвига по функции хорошего суффикса*/
void preBmGs(unsigned char *x, int m, int bmGs[]) {
int i, j, suff[XSIZE];
suffixes(x, m, suff);
for (i = 0; i < m; ++i)
bmGs[i] = m;
j = 0;
for (i = m - 1; i >= 0; --i)
if (suff[i] == i + 1)
for (; j < m - 1 - i; ++j)
if (bmGs[j] == m)
bmGs[j] = m - 1 - i;
for (i = 0; i <= m - 2; ++i)
bmGs[m - 1 - suff[i]] = m - 1 - i;
}
char * search(unsigned char *y, unsigned char *x, int m) {
int i, j, bmGs[XSIZE], bmBc[SIGMA], n;
char * yy;
yy=y;
n=strlen(y);
/*Подготовительные вычисления*/
preBmGs(x, m, bmGs);
preBmBc(x, m, bmBc);
/*Поиск*/
j = 0;
while (j <= n - m) {
for (i = m - 1; i >= 0 && x[i] == y[i + j]; --i);
if (i < 0)
return yy+j;
else
j += MAX(bmGs[i], bmBc[y[i + j]] - m + 1 + i);
}
return NULL;
}
Турбо-алгоритм Бойера-Мура
Данный алгоритм был разработан в 1994 году [34] и представляет собой дальнейшее улучшение алгоритма Бойера-Мура, не дающего значительной регрессии при использовании коротких алфавитов. Этот алгоритм позволяет запоминать сегмент текста, который сошелся с суффиксом образца во время прошлой попытки, что, в случае если произошел сдвиг по хорошему суффиксу, дает два преимущества:
· возможность перескочить через этот сегмент;
· возможность применения «турбо-сдвига» (рис 2.9.).
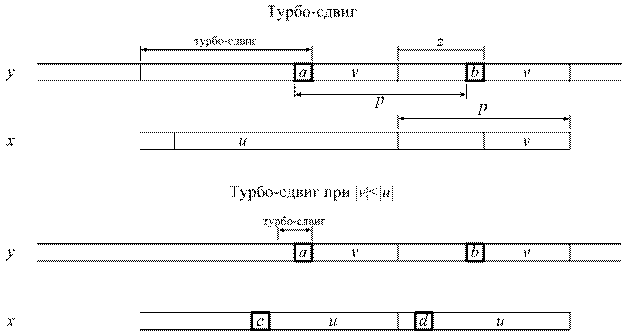
Рисунок 2.9. Осуществление турбо-сдвига.
Турбо-сдвиг может произойти, если мы обнаружим, что суффикс образца, который сходится с текстом, короче, чем тот, который был запомнен ранее.
Пусть u - запомненный сегмент, а v – суффикс, совпавший во время текущей попытки, такой что uzv - суффикс x. Предположим два символа а и b встречаются на расстоянии p в тексте, и суффикс x длины | uzv | имеет период длины p, а значит не может перекрыть оба появления символов а и b в тексте. Наименьший возможный сдвиг имеет длину | u | - | v |, который и получил название турбо-сдвига.
При | v | < | u |, если сдвиг по плохому символу больше, то совершаемый сдвиг будет больше либо равен | u | + 1. В этом случае символы c и d различны, так как мы предположили, что предыдущий сдвиг был сдвигом хорошего суффикса. Тогда сдвиг больший, чем турбо-сдвиг, но меньший | u | + 1 совместит c и d с одним и тем же символом v. Значит, если сдвиг плохого символа больше, то мы можем применить сдвиг больший, либо равный | u | + 1. Исходный код реализации турбо-алгоритма Бойера-Мура имеет следующий вид:
/*Построение таблицы для сдвига по функции плохого символа*/
void preBmBc(unsigned char *x, int m, int bmBc[]) {
int i;
for (i = 0; i < SIGMA; ++i)
bmBc[i] = m;
for (i = 0; i < m - 1; ++i)
bmBc[x[i]] = m - i - 1;
}
void suffixes(unsigned char *x, int m, int *suff) {
int f, g, i;
suff[m - 1] = m;
g = m - 1;
for (i = m - 2; i >= 0; --i) {
if (i > g && suff[i + m - 1 - f] < i - g)
suff[i] = suff[i + m - 1 - f];
else {
if (i < g)
g = i;
f = i;
while (g >= 0 && x[g] == x[g + m - 1 - f])
--g;
suff[i] = f - g;
}
}
}
/*Построение таблицы для сдвига по функции хорошего суффикса*/
void preBmGs(unsigned char *x, int m, int bmGs[]) {
int i, j, suff[XSIZE];
suffixes(x, m, suff);
for (i = 0; i < m; ++i)
bmGs[i] = m;
j = 0;
for (i = m - 1; i >= 0; --i)
if (suff[i] == i + 1)
for (; j < m - 1 - i; ++j)
if (bmGs[j] == m)
bmGs[j] = m - 1 - i;
for (i = 0; i <= m - 2; ++i)
bmGs[m - 1 - suff[i]] = m - 1 - i;
}
char * search(unsigned char *y, unsigned char *x, int m) {
int bcShift, i, j, shift, u, v, turboShift, bmGs[XSIZE], bmBc[SIGMA], n;
char * yy;
yy=y;
n=strlen(y);
/*Подготовительные вычисления*/
preBmGs(x, m, bmGs);
preBmBc(x, m, bmBc);
/*Поиск*/
j = u = 0;
shift = m;
while (j <= n - m) {
i = m - 1;
while (i >= 0 && x[i] == y[i + j]) {
--i;
if (u!= 0 && i == m - 1 - shift)
i -= u;
}
if (i < 0)
return yy+j;
else {
v = m - 1 - i;
turboShift = u - v;
bcShift = bmBc[y[i + j]] - m + 1 + i;
shift = MAX(turboShift, bcShift);
shift = MAX(shift, bmGs[i]);
if (shift == bmGs[i])
u = MIN(m - shift, v);
else {
if (turboShift < bcShift)
shift = MAX(shift, u + 1);
u = 0;
}
}
j += shift;
}
return NULL;
}
Алгоритм Чжу-Такаоки
Этот алгоритм, разработанный в 1987 году [73], представляет собой еще одну реализацию алгоритма Бойера-Мура, специально оптимизированную под короткие алфавиты. Эго основной целью было улучшение эвристики функции плохого символа, которая у обычного алгоритма Бойера-Мура при использовании таких алфавитов отказывала уже на коротких суффиксах. При этом улучшение эвристики достигается благодаря тому, что вместо таблицы для одного плохого символа, этот алгоритм строит таблицы для пары символов, несовпавшего и идущего перед ним. Данная таблица представляется в виде матрицы ztBc размера [SIGMA] * [SIGMA]. Исходный код реализации алгоритма Чжу-Такаоки имеет следующий вид:
void suffixes(unsigned char *x, int m, int *suff) {
int f, g, i;
suff[m - 1] = m;
g = m - 1;
for (i = m - 2; i >= 0; --i) {
if (i > g && suff[i + m - 1 - f] < i - g)
suff[i] = suff[i + m - 1 - f];
else {
if (i < g)
g = i;
f = i;
while (g >= 0 && x[g] == x[g + m - 1 - f])
--g;
suff[i] = f - g;
}
}
}
/*Построение таблицы для сдвига по функции хорошего суффикса*/
void preBmGs(unsigned char *x, int m, int bmGs[]) {
int i, j, suff[XSIZE];
suffixes(x, m, suff);
for (i = 0; i < m; ++i)
bmGs[i] = m;
j = 0;
for (i = m - 1; i >= 0; --i)
if (suff[i] == i + 1)
for (; j < m - 1 - i; ++j)
if (bmGs[j] == m)
bmGs[j] = m - 1 - i;
for (i = 0; i <= m - 2; ++i)
bmGs[m - 1 - suff[i]] = m - 1 - i;
}
/*Построение таблицы для сдвига по функции плохого символа*/
void preZtBc(unsigned char *x, int m, int ztBc[SIGMA][SIGMA]) {
int i, j;
for (i = 0; i < SIGMA; ++i)
for (j = 0; j < SIGMA; ++j)
ztBc[i][j] = m;
for (i = 0; i < SIGMA; ++i)
ztBc[i][x[0]] = m - 1;
for (i = 1; i < m - 1; ++i)
ztBc[x[i - 1]][x[i]] = m - 1 - i;
}
char * search(unsigned char *y, unsigned char *x, int m) {
int i, j, ztBc[SIGMA][SIGMA], bmGs[XSIZE], n;
char * yy;
yy=y;
n=strlen(y);
/*Подготовительные вычисления*/
preZtBc(x, m, ztBc);
preBmGs(x, m, bmGs);
/*Поиск*/
j = 0;
while (j <= n - m) {
i = m - 1;
while (i >=0 && x[i] == y[i + j])
--i;
if (i < 0)
return yy+j;
else
j += MAX(bmGs[i],ztBc[y[j + m - 2]][y[j + m - 1]]);
}
return NULL;
}
Алгоритм GRASPM
Genomic Rapid Algorithm for String Pattern Matching (GRASPM) – алгоритм разработанный Серхио Диосдадо и Пауло Карвальо в 2009 году [35], специально для анализа геномных последовательностей. Этот алгоритм основан на использовании перекрывающегося 2-грамм частотного анализа, который позволяет улучшать эвристику сдвига. Алгоритм ориентирован на быстрый поиск коротких строк (последовательностей до 30 символов) в тексте, состоящем из ограниченного набора часто-повторяющихся символов. Исходный код реализации алгоритма GRASPM имеет следующий вид:
typedef struct GRASPmList {
int k;
struct GRASPmList *next;
} GList;
void ADD_LIST(GList **l, int e) {
GList *t = (GList *)malloc(sizeof(GList));
t->k = e;
t->next = *l;
*l = t;
}
char * search(unsigned char *t, unsigned char *p, int m) {
GList *pos, *z[SIGMA];
int i, j, k, first, hbc[SIGMA], n;
char * yy;
yy=t;
n=strlen(t);
/*Подготовительные вычисления*/
for(i=0; i<SIGMA; i++)
z[i]=NULL;
if (p[0] == p[m-1])
for(i=0; i<SIGMA; i++)
ADD_LIST(&z[