

Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...

Типы оградительных сооружений в морском порту: По расположению оградительных сооружений в плане различают волноломы, обе оконечности...
Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...
Типы оградительных сооружений в морском порту: По расположению оградительных сооружений в плане различают волноломы, обе оконечности...
Топ:
Основы обеспечения единства измерений: Обеспечение единства измерений - деятельность метрологических служб, направленная на достижение...
Отражение на счетах бухгалтерского учета процесса приобретения: Процесс заготовления представляет систему экономических событий, включающих приобретение организацией у поставщиков сырья...
Определение места расположения распределительного центра: Фирма реализует продукцию на рынках сбыта и имеет постоянных поставщиков в разных регионах. Увеличение объема продаж...
Интересное:
Национальное богатство страны и его составляющие: для оценки элементов национального богатства используются...
Аура как энергетическое поле: многослойную ауру человека можно представить себе подобным...
Берегоукрепление оползневых склонов: На прибрежных склонах основной причиной развития оползневых процессов является подмыв водами рек естественных склонов...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Данное сочетание встречается часто (1,111,174). Заболевание выявлено в различных этнических группах и географических зонах - в основном там, где распространены одновременно гены HbS и бета-талассемии, в том числе и в Дагестане.
Мы наблюдали 44 больных (44% от всех обследованных нами) в возрасте от 2 до 69 лет, которые по всем параметрам можно было отнести к S-бета-талассемии. Из них мужчинЗУ (70,¥ %), женщин 13(2¾ & %).
Критерии отнесения больных к данной группе: 1. Клиническая картина достаточно выраженная, но не соответствующая гомозиготной СКА. 2. Много мишеневидных эритроцитов в мазке периферической крови. 3. При метабисульфитной пробе не все эритроциты принимают серповидную форму. 4. Более выраженная, чем при чистой СКА спленомегалия. 5. Обнаружение при электрофорезе HbS и одновременное наличие других фракций -АиАи F. 6, Снижение ССГЭ, СрОЭ.
Уровень НЬ А позволяет не только говорить о сочетании с ГП S, но и определить тип гена талассемии (бета°или бета+).
Гематологическими критериями отнесения больных к бета0 талассемии являются: общий НЬ в среднем 80 г/л; гематокрит 25% (20%-36); HbS 50-85%; НЬА, 3,6% н HbF 20-30%.
Таких лиц из 44 наших пациентов было 9 человек.
При 5/бета+талассемии в зависимости от степени угнетения синтеза бета цепочек могут наблюдаться разные количества НЬА.
Гематологическими критериями отнесения больных к данной группе являются: общий НЬ 80-130 г/л; гематокрит 32% (25%-40%); HbS 55-75%; НЬА 15-30%; НЬА.,3,6 и HbF 1-20%.
Из наших 44 пациентов S/бета-талассемии 35 человек можно было отнести к данной группе. Подразделять их на подтипы оказалось сложно. Поэтому нами все они были отнесены к единой группе S/бета''талассемии, как, по сути дела, это делается во всем мире.
|
|
Кроме того, известны случаи бета-талассемии, протекающие со значительным повышением концентраций НЬА. Так, СВ. Колодей (8) приводит
111
значения содержания НЬА, составляющие в среднем 42,5±1,57%. Такие талассемии называют бета++-алассемиями. Соответственно, сочетание их с S будет называться Б/бета++-талассемия.
Клиническая картина сочетанных форм можетбьпъ различной - от напоминающих чистую СКА, до стертых, малосимптомных. Наиболее выражено и тяжело протекают формы 8/бета°-талассемии, но описаны и случаи тяжелого течения S/бета+талассемии у тех, чья болезнь манифестирует в детстве и достаточно выражена.
Симптоматика наших больных состояла в жалобах на общую слабость, которые предъявляли 39,2% больных, боли в костях и суставах 21,4%; болевой синдром в животе отмечался в анамнезе почти у половины больных (42,8%), увеличение печени выявлено у 28,5%, селезенки у 21,4%; изменение черепа (в виде башенного) у 1%, желтушность кожных покровов у 3,5%. койлонихии - у 10,7%.
Интересно отметить высокое содержание HbS в данной группе, который превышает таковой у гетерозиготных носителей S. Этот признак может быть использован как маркер дифференциации между чистыми формами S талассемии и смешанными вариантами. При последних вариантах серповидный гемоглобин преобладает над А. Правда, поскольку HbS плотно примыкает на электрофореграммах к НЬА, разделение этих фракций может встретить трудности. В таких.случаях может помочь генеалогический анализ и обследование членов семьи. Разумеется, что диагнозу поможет исследование темпа синтеза бета цепей у таких больных.
Проявление болезни было достаточно выраженным. У большинства обследованных лиц этой группы отмечалась анемизация - НЬ 95±4,3 г/л, эритроциты 4,1 ±0,01х10|г/л, ССГЭ 1,34±0,037 фм, СрОЭ 80,4±1,7 фл.
Как говорилось, дифференциации чистой S формы и сочетанных поражений может помочь анализ морфологии эритроцитов.
По нашему опыту можно сказать, что при сочетанных формах параллельно степени анемизации идет увеличение патологических форм эритроцитов, характерных для СКА в сочетании с бета-талассемией. Мишеневидные эритроциты составляли от 28 до 42% в поле зрения. В периферической крови выделялись нормоциты в количестве 3-6 клеток на 100 эритроцитов. Более выражен количественный микроцитоз, обнаруживались анулоциты, шлемовидные клетки. Подобные сведения можно найти и в литературе (89).
|
|
Проба с метабисульфитом натрия выявила серповидные эритроциты, они появились уже через 20-30 мин., достигая максимума через сутки. Практически 80% эритроцитов имели в это время серповидную форму.
112
Тест на растворимость НЬ был положительным у 100% обследованных, помутнение раствора произошло на 3 минуте. У 75% больных этой группы наблюдалось увеличение содержания фетального и А2 НЬ.
Средний уровень сывороточного железа колебался в пределах нормы (17,3±0,5 мкмоль/л), хотя у ряда больных и имелись признаки железо-дефицита (койлонихии).
Следует подчеркнуть, что в Дагестане достаточно часто у больных ГП встречаются низкие показатели железа сыворотки крови и признаки сочетания их с ЖДА.
Какое-то значение в дифференциальной диагностике имеют размеры селезенки. Как указывалось в разделе клиники, при чистом варианте СКА спленомегалия у детей наблюдается не всегда и даже может иметь место «аутоспленэктомия». При смешанных же вариантах спленомегалия наблюдается практически всегда.
При электрофоретическом исследовании гемолизатов 29 лиц с сочетанием ГП S и бета-талассемии у 9 человек НЬА полностью отсутствовал. Это свидетельствовало о том, что у данных больных имеет место сочетание S с бета°-талассемией. При последнем варианте заболевания синтез бета глобиновых цепей не происходит, НЬА не синтезируется и по этой причине, отсуствует в крови таких больных. Сочетанная форма бета°-талассемии с HbS по клиническому течению близка к СКА.
У/"человек НЬА был на уровне 26%, у 15 - количество HbS и НвА было одинаковым. Эти данные, свидетельствуют, что в Дагестане СКВ чаще сочетается с одним из вариантов В+ талассемии.
Для иллюстрации сказанного приводим описание семьи Б-х.
Пробанд Б-ов С, 11 лет, наблюдался ранее, амбулаторно, в клинике обследован в феврале-марте 1985 года, и/б №91/1191.
Болеет с младенчества. Родители отмечали приступы беспричинного плача, длившиеся иногда в течение нескольких дней и прекращавшиеся при крепком пеленании, в тепле. Рост и развитие шло медленно. Когда ребенок овладел речью, он стал жаловаться на приступообразные боли во всех суставах, возникающие периодически, несколько раз в месяц. В первые дни приступа боли были очень сильными, почти нестерпимыми, не давали больному заснуть, держались 4-5 дней и постепенно проходили. Провоцирующими факторами часто служили холод, сырость; в теплое время года приступы возникали гораздо реже, а в жаркие летние месяцы не отмечались вовсе.
|
|
113
Лечение получал амбулаторно и стационарно. До нашего обследования ставился диагноз полиартрита.
Объективно: значительно отстает в массе и росте от должного, рост 125, масса тела - 24,5. В то же время внешне резких изменений скелета, характерных для гомозиготной серповидноклеточной анемии, нет. Кожные покровы бледные, склеры глаз слегка субиктеричны. Периферические лимфоузлы не увеличены. В легких перкуторно - легочный звук, аускуль-тативно везикулярное дыхание. Границы сердца в пределах нормы, тоны ясные, чистые, пульс ритмичный, 74 в 1 мин., АД 100/70 мм рт. ст. Органы пищеварения: язык чистый, влажный, живот без особенностей, печень и селезенка не увеличены. Область почек на глаз не изменена.
Анализ крови: Нв ПО г/л, эритр. 3,6х10х|2/л, СОЭ 19 мм в час, лейкоц. 4,6x109/л, в формуле: э. 3%, п. 2%, с. 56%, л.30 % и м. 9 %. Ретикулоц. 2,5 %, тромбоц. 316,8x10%.
В мазках периферической крови: гипохромия, анизопойкилоцитоз, полихроматофильные эритроциты, нормоциты 5:100 клеток. Количественный микроцитоз 5,5%. Мишеневидных эритроцитов-более 20% и отдельные необратимо серповидные эритроциты (рис. 16).

Рис. 16. Периферическая кровь б-го Б-ва. Более 20% эритроцитов мишене-видны. Отмечается анизопойкилоцитоз, полихромазия эритроцитов, отдельные необратимо серповидные клетки (на 12 часов и в центре). Окуляр 7, объектив 90 (собственные оригинальный препарат и фото).
Билирубин в сыворотке крови 21,3 мкмоль/л (реакция непрямая), сывороточное железо 28,6 мкмоль/л, холестерин сыворотки 3,32 ммоль/л, бета липопротеиды 280 единиц.
114
Натрий сыворотки 134,8 ммоль/л, калий 4,09 ммоль/л, кальций 2,14 ммоль/л; сахар крови 5,9 ммоль/л: сиаловые кислоты 0,90. С-реактивный белок не обнаружен, протромбиновый индекс 83,3.
|
|
ОРЭ повышена (скрининг проба ++), ССГЭ 1,89 фм, СрОЭ 83 фл, СДЭ 7,2 мкм, СТЭ 2,0 мкм, СИ 3,6.
При проведении метабисульфитной пробы выявляется серповидность части эритроцитов (рис. 17).
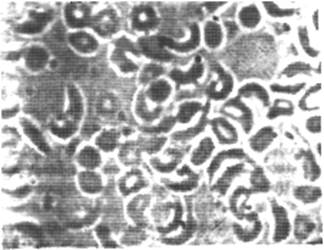
Рис.17. Серповидность при метабисульфитной пробе. Окуляр 7, объектив 90 (собственные оригинальный препарат и фото). Серповидную форму приняли не все эрироциты. Видны гиперхромные микроциты, обычные эритроциты.
Анализ мочи: относительная плотность 1010, осадок без изменений.
Рентгенологическое исследование: органы грудной клетки без патологии, при боковой рентгенограмме черепа костномозговые изменения не определяются; на рентгенограммах таза и тазобедренных суставов патологические изменения не выявляются, головки бедренных костей не деформированы, на рентгенограммах костей голени и плеча в двух проекциях патологические изменения не отмечаются.
Консультация окулиста: визус нормальный, со стороны сред глаз изменений нет, глазное дно в пределах нормы.
115

Рис. 18. Электрофореграммы больных семьи Б-вых. У спи (Б-ов Г.) S гемоглобин не выявляется. У остальных членов семьи имеется дополнительная фракция, соответствующая Hb S (собственные оригинальная Э-фореграмма и фото).
Электрокардиографическое исследование - возрастная норма. При определении фетального НЬ методом щелочной денатурации его количество составило 8%. При алектрофоретическом исследовании на ацетатной пленке содержание KbS равнялось 65%, А^ - 14%,А-13% (рис.18).
Сестра пробанда - Б-ва С, 18 лет, поступила в клинику 20 февраля 1985 года, и/б 90/1190. До 7- 8 лет развивалась нормально, без каких-либо болевых кризов. С этого возраста без всякой видимой причины стали беспокоить примерно такие же приступы болей в суставах, как и у брата. Как правило, боли наблюдаются зимой, при острых респираторных заболеваниях, переохлаждении. В жаркое время года приступов болей не отмечает. Бывала в горах, на высоте примерно 1500 метров, какого-либо ухудшения состояния не отметила. Еще года 4 назад находили снижение гемоглобина.
Жалуется на бол и во всех суставах, костях рук, ног, понижение аппетита, общую слабость, утомляемость.
Объективно: несколько отстает в развитии: рост 153, масса тела 48 кг. Кожные покровы и видимые слизистые оболочки бледные, с желтушным оттенком. Периферические лимфоузлы не увеличены. Границы сердца в пределах нормы, тоны ясные, чистые.
116
Пульс 90 ударов в 1 минуту, ритмичный, АД 100/70 мм рт. ст. Печень не увеличена, селезенка выступает из-под левого подреберья на 5 см, плотноватой консистенции, болезненная.
В анализах: общий анализ крови: НЬ 90 г/л, эритр. 2,9х10|2/л, ЦП 0,9. Лейкой, 3,3х109/л, формула: э. 1%, п. 4%, с. 51%, л. 37°/о и м. 7%. СОЭ -30 мм/час, ретикулоц. - 1,9%, тромбоц.86х109/л
|
|
В мазках периферической крови: анизо-пойкилоцитоз, иормоциты 1: 100 клеток, единичные мишеневидные клетки в каждом поле зрения. Количественный микроцитоз 6%. Метабисульфитная проба на серповид-ность положительная.
Билирубин в сыворотке крови: общий 29,9, прямой 4,27, непрямой 25,6 мкмоль/л, сывороточное железо 16 мкмоль/л, холестерин сыворотки 4,43 ммоль/л, беталипопротеиды 330 ед. Натрий сыворотки 139,2 ммоль/л, калий 5,11 ммоль/л, кальций 2,02 ммоль/л, магний 0,96 ммоль/л, фосфор 4,08 ед. Общий белок крови 72 г/л, альбумины 50%, глобулины: альфа 12%, бета 18% и гамма 20%.
Анализ мочи: относительная плотность 1015, осадок без патологии.
Рентгенологическое обследование: органы грудной клетки в норме, при рентгенографии черепа костные патологические изменения не выявляются.
Электрокардиография: синусовая тахикардия.
Специальные исследования: ОРЭ повышена (скрининг проба ++), минимальная резистентность 0,56, максимальная 0,20% хлористого натрия. ССГЭ 1,62 фм, СрОЭ 70,1 фл, СДЭ 6,9 мкм, СТЭ 2,7 мкм и СИ 2,5.
При электрофорезе на ацетатной пленке (рис. 18) выявлено: HbS - 65,5'%, А2- 8,3% и А - 11,2%. Фетального гемоглобина оказалось 15%.
Мать пробанда Б-ва 3., 43 лет. Жалоб не предъявляет. Обследована в клинике вместе с детьми. Роста и развития нормального, каких-либо заболеваний у себя не помнит, кроме обычных острых респираторных инфекций и однократной пневмонии. Со стороны внутренних органов отклонений от возрастной нормы нет.
Общий анализ крови: НЬ 160 г/л, эритр. 4,6x10|2/л, СОЭ 16 мм/час. Лейкоц. 5,9x10%, формула: э. 2%, п. 1%, с. 63%, л. 30% и м. 4%. Ретикулоц. 0,2%, тромбоц. 273x109/л.
В мазках периферической крови: анизо-пойкилоцитоз, количественный микроцитоз 6,5%.
Билирубин сыворотки крови 8,55 мкмоль/л; сывороточное железо 14,3 мкмоль/л; натрий 126,1 ммоль/л, калий 3,84 ммопь/л, кальций 2,02 ммоль/ л, магний 0,94 ммоль/л, фосфор 2,28 ед. Анализ мочи без патологии.
117
Рентгенологически костно-суставная патология не выявляется.
ЭКГ в норме; консультация невропатолога и окулиста - без отклонений от возрастной нормы.
При специальном обследовании: скрининг проба на осмотическую резистентность отрицательная, ССГЭ 1,88 фм, СрОЭ 80 фл, СДЭ 7 мкм, СТЭ2 мкм, СИ 3,5. При метабисульфитной пробе в периферической крови выявляются серповидные эритроциты.
Электрофорез на ацетатной пленке (рис.18): HbS - 39,2%, А, - 5,3'% и А - 48,5%. Фетальный НЬ по методу щелочной денатурации 7%.
Отец пробанда Б-ов Г., 42 года, обследован амбулаторно. Никаких жалоб не предъявляет, со стороны внутренних органов отклонения от нормы в момент осмотра не выявляются.
Из анализов: скрининг проба на повышение осмотической резистентности эритроцитов резко положительна; при электрофорезе (рис.18): А -75,4%, А2- 12,5%. Оставшийся 1,6% приходился на долю фетального НЬ.
В семье имеется еще трое детей - две девочки и один мальчик. Еще один ребенок умер сразу после рождения. Причина смерти неизвестна.
Родословная данной семьи представлена на схеме 3. Как следует из нее. отец имеет аномальный ген по бета"1"- талассемии, мать по В+- талассемии и HbS. Пробанд и его сестра являются компаундами - имеют качественную и количественную аномалию бета гена - Вт и HbS.

Схема 3. Родословная семьи Б-х (собственная схема).
Следует отметить, что в ряде случаев возникают серьезные трудности в дифференциации чистой СКА и сочетанных форм. Дня иллюстрации сказанного приводим собственное наблюдение.
Больная М., 48 лет, была направлена в клинику с диагнозом ревматоидного полиартрита, с жалобами на боли в костях, преимущественно в левом
тазобедренном суставе, усиливающиеся при физической нагрузке и перемене погоды. Этими болями страдает с детских лет, левая нога постепенно стала короче правой, изменилась походка.
Проводившаяся терапия индометацином и стероидными гормонами в амбулаторных условиях оказалась неэффективной.
Из анамнеза жизни: родители умерли 30 лет назад. Брат, живущий в другой республике, страдает болями в костях. Больная - одинокая женщина, детей не имеет. Менструальный цикл не нарушен. Кровопотерь не было. Заболевания желудочно-кишечного тракта отсутствуют.
Объективно: больная среднего роста, пониженного питания. Кожные покровы смуглые. Суставы, за исключением тазобедренного, не изменены. Отмечается укорочение левой ноги на 2 см, из-за чего больная прихрамывает, и значительная болезненность в тазобедренном суставе при сгибании, разгибании и ротации кнаружи. Органы дыхания, брюшной полости, мочевыделения без особенностей. Границы сердца расширены в обе стороны на 1 см, тоны приглушены. На верхушке сердца - систолический шум функционального характера. Пульс ритмичный, частота его 86 ударов в минуту. Печень у края реберной дуги. При пальпации в проекции желчного пузыря отмечается болезненность.
В анализах: НЬ 84 г/л, эритр. 3,9x10%!, гематокрит 31 объ.%, ССГЭ 1,33 фм, СрОЭ 79 фл. Морфология эритроцитов: полихрамотофилия, единичные мишеневидные эритроциты, анизоцитоз. ОРЭ минимальная 0,5%, максимальная 0,31% раствора хлористого натрия. Количество ретикулоц. 1,2%, тромбоц. 240x10%, лейкоц. 6,6х109л. Лейкоцитарная формула без изменений. Проба на серповидность положительная (рис. 19).

Рис.19. Метабисульфитная пробау б-й М. Все эритрощпы приняли серповидную форму. Окуляр 7, объектив 90 (собственные оригинальная Э-фореграмма и фото).
119
Исследование состава Hb: Hb F - 2,2%, на электрофореграммах определяется фракция аномального гемоглобина, злектрофорети че екая подвижность которой соответствует HbS (рис. 20). В элюатах концентрация HbS составляет 56%, НвА2 3,2% и НвА 39,6%. Оставшиеся 1,2%, по-видимому, приходятся на малые фракции (электрофорез у данной больной производился также и в НИИ гематологии в Москве).

Рис.20. Электрофореграмма б-й М. Четко видна большая патологическая фракция HbS между Аг и А (собственные оригинальная Э-фореграмма и фото).
Проба растворимости на присутствие HbS оложительная, сывороточное железо 23,2 мкмоль/л, сывороточные гаптоглобины 960 мг/л, сидероциты 3%, билирубин 16 мкмоль/л.
Активность фермента Г6ФД нормальная.
При рентгенологическом исследовании отмечается остеопороз костей черепа с расширением диплоид; поражение головки левой бедренной кости по типу болезни Пертеса (аваскулярный некроз).
По многим клинико-гематологическим признакам в данном случае речь должна идти о гомозиготной форме СКА (типичные болевые кризы, изменения в костях, резко положительная проба на серповидность, 56% HbS и т. д.). Однако такому диагнозу противоречит возраст больной - 48 лет, проявление болезни после 30 лет и наличие достаточно большой фракции гемглобина А при электрофоретическом исследовании. Это заставляет думать о сочетании ГП S с другой гемоглобинопатией возможно, с бета-талассемией. Однако фракция А, также не увеличена. Возможно, что речь идет о сочетании с бета°-талассемией.
В таких случаях окончательное решение вопроса требует изучения темпа синтеза глобиновых цепей. Но это не снимает вопроса о том, почему болезнь
120
у нее проявилась в таком позднем возрасте. Возможно, что у части больных СКА один из аллельных генов (комплементарный) эпистатичен. И тогда болезнь проявляется в позднем возрасте.
Генеалогический анализ или обследование родственников у данной одинокой женщины не были возможны.
СОЧЕТАНИЕ С АЛЬФА ТАЛАССЕМИЕЙ
Группу СКА в сочетании с альфа талассемией составило 32 больных -25 взрослых (18 женщин и 7 мужчин) и 7 детей (5 мальчиков и 2 девочки). Дети были в возрасте от 8 до 13 лет.
Все больные этой группы активны. В клинику госпитализировано 10 больных. В анамнеза у 15 пациентов - редкие серповидные кризы.
Жалобы: боли в суставаху 16 (66,7%) из 24, в костях нижних конечностей у 4 (16,7%), в спине и пояснице у 2 (8,3%); реакция на погоду у 5 (20,8%), слабость у 7 (29,1 %), головокружение у 4 (16,7%).
Все больные имели астеническое телосложение. Бледность кожных покровов у 13 (46,2%), причем у 7 (21,8%) она сочеталась с желтушностью. Болезненность костей и суставов при пальпации отмечалась у 3 (9,3%), высокое твердое небо у 5 (15,6%).
Сердечно-сосудистая система: тахикардия у 10 и систолический шум у 4 больных. На ЭКГ у 4 пациентов синусовая аритмия. Умеренная сплено-мегалияу 2, агепатомегалия-у 5.
У одной больной выявлен хронический пиэлонефритс нарушением концентрационной функции почек.
Гематологическими критериями отнесения больных к данной группе являются: HbS <40%; НЬА 50-55%; HbAjB норме.
Гематологическая картина наших пациентов: Hb 102±2,9 г/л, эритр 3,95±0,12х10|2/л, гематокрит 36,4±1,0 объ.%. Эритроцитарные показатели: СрОЭ 89,5±2,5 фп, ССГЭ 0,38 фм, СКГЭ 25±0,9%. Лейкоц. 5,8±0,67х107л, ретикулоц. 1,7±0,5%.
В мазках периферической крови у всех больных отмечались анизо-пой-килоцитоз, серповидные и мишеневидные эритроциты, у 6 - микроцитоз, гипохромия.
Проба на серповидность положительна у всех больных. Электро-форетически: HbS 30,6±0,95%, НЬА2 2,17*0,11%, HbF 1,6*0,12% и НЬА 65,9*1,06%.
Общий белок в сыворотке (10 больных) 73,5±1,6г/л; билирубин (30 больных) 15,3±0,7 мкмоль/л.; сывороточное железо (9 больных) 8,4±0,39 мкмоль/л.
121
В литературе также описаны случаи сочетания СКА с альфа-талассемией (6,87,98,173,186 и др.). При отсутствии возможности определеить темп синтеза альфа цепей о сочетании судят по наличию признаков S/Th, которая не оказывает ухудшающего влияния на течение болезни. Такая «не взаимо-действующейая» талассемия считается вероятной формой альфа-талассемии. Еще одним признаком может служить то, что при сочетании гомозиготной формы ГП S и альфа-талассемии уровень HbS ниже, чем ему положено быть,
Сочетание СКА с альфа-талассемией в большинстве случаев протекает в менее тяжелой форме, чем в чистой форме.
A. von Enk et al. (181) описали сочетание СКА с альфа-талассемией у 2 женщин африканского происхождения. Заболевание протекало доброкачественно, у них неоднократно наступала беременность, которая прошла без осложнений.
Электрофорез НЬ выявлял присутствие HbS (82-84%), HbF (12 - 15%), HbAj(3%) и следы Hb Bart.
Обычно доброкачественное течение сочетания ГП S и альфа-та-лассемии объясняют повышенным уровнем HbF, который имеет место при этом. Есть и другие объяснения данному явлению.
Считается, что в крови таких больных образуются гибридные молекулы типа S|6eTas|raMMa, которые снижают возможность полимеризации HbS.
Имеет значение и количество HbS в крови больных. На большом материале Э.Г. Гаджиев и соавторы (6) нашли, что, если при сочетании ГП S с альфа-талассемией HbS в крови ниже ЗОУо - болезнь протекает лишь с клиническим проявлениями, но без гематологических нарушений. Если же HbS выше 38%, то у таких больных возникают и гематологические нарушения.
Иногда наслоение альфа-талассемии не вызывает существенных изменений симптоматики СКА (128).
В литературе имеются описание и других вариантов сочетания СКБ сальф-талассемией. Они показывают, что клиника болезни может быть различной в зависимости оттого, сколько альфа цепочшвых гена являются дефективными.
Одна из наблюдаемых нами семей предсталяет чрезвычайный интерес для генетики. Пробанд и один из сибсов (сестра) были обследованы не только в нашей клинике, но и в бывшем ЦНИИГиПК. Данные по этой семье приводятся ниже.
Пробанд А-ва Г., 3 8 лет, поступала в клинику пропедевтики внутренних болезней Дагмедакадемии неоднократно, начиная с 1986 г., когда ей было 19 лет. Кроме того, она была обследована и в Москве.
Жалобы на периодически усиливающиеся боли в левом подреберье, в поясничной области слева, в мышцах, костях, общую слабость.
122
Болеет с дегства, периодически беспокоили боли в левом подреберье. Неоднократно лечилась в детских стационарах с диагнозом анемия. Всегда отмечали увеличенную селезенку. Чем лечили, не знает, эффекта от лечения не было.
Детскими инфекциями не болела. Часто отмечает простудные заболевания. Менструации с 20 лет. Имеет 3-х братьев и 1 сестру, у одного из братьев имеется дефект зрения (выявлен микрофтальм), родители считают себя здоровыми.
Больная пониженного питания, отстает в физическом развитии. Масса тела 38,5 кг, рост 153 см. Кожные покровы бледные, склеры слегка икте-ричны. Со стороны костно-мышечной системы деформаций нет.
В легких аускультативно везикулярное дыхание, перкуторно легочный звук. ЧДД 17 в мин.
Тоны сердца приглушены, 1 тон ослаблен, выслушивается дующий систолический шум по левому контуру сердца во 2-3-4 межреберьях. Первый тон на верхушке сохранен. Пульс 100 в мин., АД 120/70 мм рт.ст.
Язык чистый, влажный, отмечается высокое небо; живот увеличен в размерах, напряжен. Печень увеличена за счет правой доли на 4-5 см, край плотный. Увеличение селезенки видно на глаз, она выступает из-под реберной дуги на 14-15 см, болезненна. Стул оформленный, регулярный.
Область почек на глаз не изменена. Мочеиспускание свободное, безболезненное.
Анализы: НЬ 110 г/л, ЦП0,8,эритр. 2,93х10|2/л, лейкоц. 7х] 07л;формула: п. 9%; с. 55%; л.35%, м. 1%. СОЭ 22 мм/ч. Тромбоц. 136x107л. В мазке -гипохромия ++, анизоцитоз +++, микроцитоз, мишеневидные эритроциты значительное количество (рис. 21).
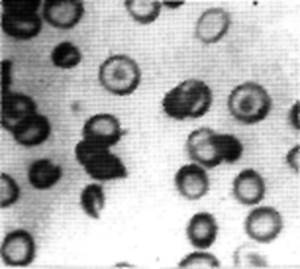
Рис. 21. Видно значтительное количество мишеневидиых клеток, некоторые клетки с разрывом и склонностью к серповидности, есть грушевидные эритроциты. Окуляр 7, объектив 90 (собственные оригинальные препарат и фото). 123
Ретикулоц. 2,2%, гематокрит25 обь.%. Ср.ОЭ 71 фм, СТЭ2,1 мкм, СДЭ 5,5 мкм, ССГЭ 27,4 пг.
Метабисульфитная проба выявляет серповидность (рис. 22).

Рис. 22. Серповидность эритроцитов при метабисульфитной пробе. Видно, что часть эритроцитов приняли серповидную форму, но большая часть осталась нормальной. Окуляр 7, объектив 90 (собственные оригинальные препарат и фото).
Билирубин 29,9 мкмоль/л, непрямой25,6, прямой 4,3. Общий белок 95 г/л. Сывороточное железо 20 мкмоль/л. ОЖСС 82 мкмоль/л. Г-6ФД в норме. Мочевина крови 3,46 мкмоль/л; Na 156,6 ммоль/л, К 3,58 ммоль/л, Са 2,32 ммоль/л, Mg 1,04 ммоль/л, фосфор 3,06 ед. Бета-липопротеиды 370 мг%. ACT 0,58 мкмоль/л, АЛТ 0,78 мкмоль/л, ЩФ - 2,59 ммоль/л. lgG 14,0; IgA 2,1; IgM 1,35. Гагттоглобин 1,0; трансферрин 2,9; церулоплазмин 0,45; ЦИК-690. Ферритин сыворотки крови 900 мкг/л, ферритин гемолиз. 87,1 мкг/л. Электрофорез гемоглобина: HbS 88,8%, HbF 11,2%, Hb Барт следы. ОРЭ (-Н-). Проба на внутри клеточные включения по ложит ельна» (рис. 23).

Рис. 23. Внутриклеточные включения при БКС пробе. Окуляр 7, объектив 90 (собстенные оригинальный препарат и фото).
УЗИ от 3.11.86 г.- печень увеличена на 4 см, паренхима диффузно уплотнена с точечными кальцификатами, сосудистый рисунок выражен. желчные долевые ходы сохранены, несколько сужены. Желчный пузырь грушевидной формы, крупный малоподвижный 2,7-3-9,4 см с плотными отечными стенками, толщиной до 0,6 см. В проекции пузыря - массаэхоплот-ных включений размерами от 1,3 до 1,8 см. Почки одинаковых размеров 5,5-5,7-10,6 см, контуры ровные. Паренхима однородная. Собирательная система (без водной нагрузки) расширена. В проекции чашечек мелкие эхо-плотные включения в виде теней и песка. Заключение: гепатохолецистит. Желчнокаменная болезнь. Пиелонефрит.
Нами обследованы еще 3 сибса. У всех у них положительна метабисульфитная проба, у двух в эритроцитах обнаружены внутриэритроцитарные включения, у одного дефицит Г6ФДГ и у одного повышен уровень А2. Родословная семьи представлена ниже (схема 4).

Схема 4. Родословная семьи А-ых (собственная схема)
Пояснения к родословной: а - нормальный альфа цепочковый ген; а - аномальный альфа цепочковый ген: р - нормальный бета цепочковый ген; р+ - аномальный бета цепочковый ген; Г6ФДГ - ген дефицита ГбФДГ; S - ген серповидного гемоглобина.
Таким образом, семья имеет чрезвычайно сложную генетическую патологию.
Пробанд: носитель гена сероповидного гемоглобина и аномальных альфа цепочковых генов.
Отец имеет один аномальный альфа цепочковый ген, аномальный ген по Г6ФД и ген серповидного гемоглобина.
Мать имеет один аномальный альфа цепочковый ген, аномальный (3+ ген и ген серповидного гемоглобина.
125
Первый сибс имеет аномальный альфа цепочковый ген, аномальный В+ ген и ген серповидного гемоглобина.
Второй сибс - аномальный альфа цепочковый ген, 3+ ген, ген дефицита Г6ФДГ и серповидного гемоглобина.
Другой сибс - имеет аномальный альфа цепочковый ген и ген серповидного гемоглобина.
У пробанда, несмотря на наличие в крови 11,8% HbF заболевание имеет выраженные проявления. Такие случаи описаны и в литературе.
Описаны сочетания ГП Бис другими формами болезни - с D, С, Е, наследственным персистированием фетального гемоглобина (НПФГ) и др. В Дагестане нами описан гемглобин D-Пеиджаб (24). Однако он встречается л ишь у ногайцев — выходцев из Индии, которые не подвержены FTIS. Кроме того, эти формы ГП и в других странах встечаются не так уж часто. В связи с этим эти редкие формы нами здесь не приводятся.
СОЧЕТАНИЕ С ЖДА
Мы наблюдали 19 пациентов, имеющих сочетание HbS с ЖДА, из них 8 мужчин (42,1%) и 11 женщин (57,9%). Основное заболевание было представлено следующим образом: гомозиготы SS - 3, S/b'TTi - 5, S/B°-th -I, AS гетерозиготы - 2, S/альфа талассемия - 8).
Приводим историю болезни одной из пациенток.
Больная 3., 42 лет, азербайджанка, предъявляет жалобы на слабость, повышенную утомляемость, незначительные боли в костях, преимущественно трубчатых, возникающие иногда при респираторных заболеваниях, перемене погоды и т. д.
Указанные проявления болезни отмечаются с детских лет, Объективно; состояние больной удовлетворительное. Телосложение правильное. Кожные покровы и видимые слизистые бледные, сухие. Костно-мышечная система без особенностей. Волосы на голове сухие, ломкие.
Отмечаются выраженные койлонихии (рис. 24).
126

Рис. 24. Выраженные койлонихии у б-й 3. (собственное оригинальное фото).
Зубы редкие. Тоны сердца ритмичные, на верхушке и над легочной артерией выслушивается систолический шум. АД -110/80 мм рт. ст. Печень увеличена, выступает из-под реберной дуги на 2 см, безболезненная, селезенка пальпируется у края реберной дуги.
Из анамнеза жизни выяснилось, что у больной было 13 беременностей, из которых 5 закончились медицинскими абортами. Из 8 родившихся детей два ребенка умерли до года. Менструальный цикл регулярный. Продолжительность менструаций 6 дней, в первые 2 дня обильные.
Гематологические исследования: НЬ 100 г/л, эритр. 4,5x10х'7л, гематокрит 0,39 объ%, ССГЭ 1,37 фм, СрОЭ 86 фл, СДЭ 6,8 мкм; количество ретикулоц. 1,2%. Морфология эритроцитов - анизопойкилоцитоз, единичные мишеневидные клетки, полихромия с преобладанием гипохромии.
Проба на серповидность эритроцитов с метабисульфитом натрия положительная. ОРЭ нормальная. Проба растворимости на присутствие HbS положительная.
Исследование состава Hb: HbF 1,2%, НЬА2 2,1%, HbS 32%, НЬА 64,7%. Сывороточное железо 10 мкмоль/л, гаптоглобины 1100 мг/л. Сидероциты отсутствуют. Билирубин крови 11,9 мкмоль/л, общий белок 70 г/л, белковые фракции: альбумины 56%, альфа, глобулины 6,0%, альфа^ глобулины 9,3%, бета глобулины 11,8%, гамма глобулины 17%. Тимоловая проба 4 ед., холестерин 6,6 м моль/л, альдолаза 5 ед., лактатдегидрогеназа 300 ед., протромбин 94%.
Пример показывает, что у больной имеется сочетание серповиднокле-точного носительства с ЖДА. Клиническое течение при таком сочетании не тяжелое, болевые проявления выражены незначительно.
127
Сочетание ЖДА с различными формами ГП приводит к различным фенотипическим проявлениям. В одних случая* (бета талассемия + ЖДА) внешнесредовый и генетический факторы действуют суммарно, ухудшая общее состояние и обусловливая специфический симптомокомплекс. В других (HbS + ЖДА) отмечается смягчение наследственной болезни на фоне железодефицитного состояния.
ЗАКЛЮЧЕНИЕ
Итак, СКБ - тяжелая молекулярная патология, наследуемая аутосомно-рецессивно и кодоминантно, вследствие чего у гомозигот может развиться болезнь различной степени экспрессивности.
В России данная патология встречается у 2 групп населения. 1. У азербайджанцев, издавна компактно проживающих на территории Дагестана и других граждан, имеющих брачные связи с азербайджанцами. 2. Среди различных диаспор или отдельных лиц, легально или нелегально проживающих на территории России. Это представители различных наций и континентов: та же азербайджанская диаспора ныне широко представленная почти во всех регионах России; группа людей, живущая в стране или обучающаяся в вузах - выходцы из многих стран Азии (Индия, Пакистан, Бангла-Деш. весь Ближний и Средний Восток и т.д.), представители всех стран черной Африки, Латинской Америки, Карибского бассейна, европейских стран региона Средиземного моря, чернокожие американцы и многие др.
СКБ, по-видимому, возникла в древние времена и получила широкое распространение в регионе, известном как «малярийный пояс» земли. Такое распространение было связано с действием одного из факторов естесственного отбора. В период свирепствования тропической малярии, носительство мутантного гена Hb S оказалось активным приспособительным фактором, направленным на сохранение вида - такие лица не болели малярией или болели ею в легкой форме. Люди с нормальным генотипом болели и умирали от малярии, больные гомозиготными формами СКБ умирали от этой болезни - действовал так называемый сбалансированный полиморфизм. В результате популяции сокращались, а число гегерозигот по HbS в ней росло.
Гомозиготы по СКБ чаще начинают болеть с окончания младенческого периода. Особенно сильно они болеют в детском возрасте.
У гомозигот болезнь проявляется тяжелыми поражениями практически всех органов и систем, вызванными вазоокклюзионнымн процессами в них и анемией, обусловленной гемолизом ригидных серповидных эритроцитов.
128
Гетерозиготы являются практически здоровыми людьми, хотя при стечении ряда средовых обстоятельств и у них могут развиться легкие формы болезни.
Для уточнения распространения данного заболевания были разработаны скрининговые методы диагностики, обеспечивающие вероятностное выявление болезни в популяции и более сложные уточняющие методы, позволяющие верифицировать болезнь. Благодаря этим методам и вниманию здравоохранения к патологии к настоящему времени уточнено ее распространение в мире, многие моменты клиники, патогенеза и лечебных мер.
Достаточно часто во всех тех регионах, где обычно встречаются СКБ, распространены также и талассемии. В связи с этим нередко встречаются смешанные формы ГП - двойные гетерозиготы - сочетание СКБ и талассемии (так называемая дрепаноталассемия), которые также ведут к тяжелым поражениям многих органов и систем организма человека.
В связи с этим даже в 80-х годах XX столетия продолжительность жизни таких больных не превышала 20-22 лет.
В течение второй половины XX столетия шли интенсивные поиски по выяснению генеза многих симптомов болезни и путей воздействия на них и на суть болезни в целом. В результате было показано, что тяжесть проявлений (экспрессивность) СКБ и ее сочетаний с талассемией зависит не только от генетических факторов, от гомозиготности по патологическому гену, но и от многих других моментов. Установлено, что большую роль в ослаблении экспрессивности болезни играют индивидуально-психологические, бытовые, социальные и другие факторы. Были поняты механизмы развития многих проявлений болезни и разработаны меры воздействия на них, в лечение введен целый ряд новых средств, разработаны и внедрены в практику приемы психосоциальной реабилитации. Все это принесло свои плоды - к началу XXI столетия медиана продолжительности жизни больных СКА и дрепаноталассемией во многих странах достигла 40-42 лет, с разбросом примерно от 29 до 51 года.
Средний возраст наших больных с гомозиготной формой ГП S составил 20 лет (с колебаниями от 10 до 34 лет).
В последние годы предпринимаются также попытки прямого излечения болезни. Во-первых, это трансплантация костного мозга. В мире к 2003 году было сделано более 200 ТКМ. При удачном подборе донора (особенно, если донором является здоровый брат или сестра) по HLA-системам приживляемость и эффективность процедуры высокая. К сожалению,
129
широкому распространению ТКМ мешает его квазивысокая стоимость и связанные с этим все и всяческие нарушения медицинской этических положений, атакже высокий риск гибели больных на предоперационном подготовительном этапе. Во-вторых, более дешевым и менее опасным является трансплантация стволовых клеток. Она также производится больным СКА и дрепаноталассемией, приводит к возникновению химеризма с преобладанием аутосомных клеток организма со здоровым генетическим материалом и значительному улучшению. В третьих, выполнен ряд экспериментальных работ по пря
|
|
|

Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций...

Своеобразие русской архитектуры: Основной материал – дерево – быстрота постройки, но недолговечность и необходимость деления...
Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...

Археология об основании Рима: Новые раскопки проясняют и такой острый дискуссионный вопрос, как дата самого возникновения Рима...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!