Визуализация
При синдроме Ротмунда-Томсона рекомендуется получить рентгенограммы длинных трубчатых костей, в возрасте до 5 лет, из-за высокой частоты скелетных дисплазий, многие из которых могут быть клинически бессимптомными.
Генетическое тестирование
В результате анализа последовательности RECQL4, генетиками могут быть зафиксированы мутации. Эти мутации встречаются приблизительно у более 66% пациентов с диагнозом синдрома Ротмунда-Томсона. Несмотря на то, что чувствительность генетического теста составляет 66%, клиническая специфичность приближается к 100%. Таким образом, отрицательный результат теста не исключает диагноз синдрома Ротмунда-Томсона, но положительный тест будет подтверждающим. Однако, в классических случаях, правильный диагноз может быть поставлен только на клинических данных (например, с раннее начало развитие сыпи на лице, которая сопровождается радиально-лучевыми дефектами, дефектами роста и редкими волосами).
Синдром Ротмунда-Томсона. Лечение
Лицам с синдромом Ротмунда-Томсона следует регулярно применять солнцезащитные кремы. Кроме того, кератолитики и ретиноиды были довольно успешными в лечении гиперкератоза. Телеангиоэктазии можно лечить лазерной терапией.
Синдром Ротмунда-Томсона. Осложнения
· Остеосаркома возникает у целых 32% пациентов с синдромом Ротмунда-Томсона. Они наиболее часто возникают в большеберцовой / малоберцовой костях. Развитие остеосарком среди пациентов с этим синдромом сильно коррелирует с типом мутации в гене RECQL4.
· У некоторых лиц может развиться либо болезнь Боуэна, либо плоскоклеточный рак.
Синдром Ротмунда-Томсона. Прогноз
Шеффер
При отсутствии злокачественности, пациентам обычно не стоит сильно беспокоиться
Синонимы синдрома Шефера. S. Siemens—Schaefer. Наследственная эктодермальная полидисплазия. Определение синдрома Шефера. Врожденный дискератоз, по-видимому, идентичный S. Jadassohn—Lewandowsky. Относится к кератозам. Автор. Schafer Е.— немецкий дерматолог. Впервые синдром был описан в 1925 г. Симптоматология синдрома Шефера: 1. Ладонно-подошвенный гиперкератоз с гипергидрозом. 2. Лейкокератоз слизистой оболочки ротовой полости. 3. Распространенный фолликулярный гиперкератоз остальных участков кожного покрова с маленькими очагами рубцовой аллопеции (hypotrichosis areata). 4. Пахионихия. 5. Врожденная катаракта. 6. Общие расстройства развития (малый рост, недостаточный вес, малые размеры лицевого черепа, низкий лоб, олигофрения, крипторхизм, гипоплазия яичек и др.). Этиология и патогенез. Наследственное расстройство, видимо, идентичное S. Jadassohn—Lewandowsky. Дифференциальный диагноз. Другие формы кератоза.
Источник: https://meduniver.com/Medical/genetika/sindrom_shefera.html MedUniver
Синдром Маринеску — Шёгрена (синдром Маринеску — Драганеску — Василиу, синдром Маринеску — Шегрена — Гарленда)[1] — редкое наследственное заболевание, характеризующееся двусторонней врождённой катарактой, мозжечковой атаксией, мышечной гипотонией, умственной отсталостью, задержкой роста, костными аномалиями (кифосколиоз, арахнодактилия и др.). Тип наследования — аутосомно-рецессивный. Лечение симптоматическое. Синдром Маринеску — Шёгрена связан с мутациями гена SIL1. Заболевание получило своё название в честь румынского невропатолога Георге Маринеску (1863—1938) и шведского психиатра Торстена Шёгрена (швед. Karl Gustaf Torsten Sjögren; 1896—1974).
Синдром известен сегодня под названием «синдром Маринеску–Шегренана», но на самом деле является вариантом «синдрома Шегрена» (он описан в 1935 году и характеризуется сочетанием врожденной катаракты с олигофренией). В разработке этого синдрома сотрудничала и румынская неврологическая школа. Этиопатогенез синдрома Маринеску–Шегрена. Синдром Маринеску–Шегрена является наследственной спинно-мозжечковой дегенерацией из-за неизвестной причины. Синдром появляется в очень раннем возрасте (до года) и затрагивает приблизительно в той же мере оба пола. Передается наследственно (существуют и некоторые исключения) по аутосомно-рецессивному типу, очень редко аутосомным геном (небольшое число случаев, описанное до 1969 г. — только 50 —выступает в пользу этого предположения). Единокровие родителей, по-видимому, играет важную роль в появлении наследственной передачи синдрома. В некоторых случаях доказан и семейный характер синдрома. Генетические исследования (кариограмма) оставались отрицательными во всех исследованных случаях. Еще в 1930 году три румынских исследователя, два невролога и один офтальмолог, обратили внимание на тяжелые неврологические проявления больных с врожденной катарактой и олигофренией, а заслуга Шегрена состоит в том, что недавно, в 1950 году, он систематизировал и описал полностью характерные аномалии этого синдрома. С момента начала описания, до сих пор, этот синдром появляется в медицинской литературе под разными названиями: синдром Маринеску–Шегрена – Гарланда; синдром Маринеску — Гарланда; синдром Маринеску–Драганеску – Василиу; синдром катаракты – олигофрении — мозжечковой атаксии. Симптоматология синдрома Маринеску–Шегрена. врожденная двусторонняя катаракта. Вначале зональная, быстро становится полной, даже в течение раннего детства; начинается с рождения и проявляется обычно к 4-м годам; олигофрения, проявляющаяся клинически — поздно, от обычного слабоумия до идиотизма; спинно-мозжечковая атаксия совмещает все неврологические признаки, постоянно присутствующие в клинической картине синдрома, появляющиеся даже с первого года жизни; мозжечковая атаксия, с нескоординированными движениями и интенционным дрожанием; отсутствие равновесия при ходьбе и в стоячем положении; нистагм; гипотония мышц с пониженными рефлексами; амиотрофии (проявляющиеся поздно); различные парезы и параличи мышц. Спиномозжечковая атаксия появляется рано и всегда до развития катаракты. Иногда, неврологические проявления напоминают клиническую картину спастической диплегии или нейрональной амиотрофии (Chareot-Marie и Tooth). Другие аномалии, сочетающиеся непостоянно: глазные: внутренне перемещающееся косоглазие; двусторонний эпикантус; маятниковый нистагм; вторичная двусторонняя катаракта; аномалии скелета: кифосколиоз; лордоз; кривая нога; аномалии суставов и пальцев; аномалии придатков кожи (дистрофические ногти; ломкие волосы); аномалии половых органов (гипоспадия; эписпадия); нанизм. Диагностика синдрома Маринеску–Шегрена. Биологические обследования не выявляют характерных и постоянных изменений. Только в некоторых случаях, при электрофорезе сыворотки крови, было выявлено нарастание содержания бета, альфа 2 и гаммаглобулинов, но и оно незначительно. Электроэнцефалограмма и пневмоэнцефалограмма выявляют иногда аномалии, показывающие полную атрофию коры мозжечка или только некоторых его долей. Патологоанатомическое обследование при вскрытии показало анатомо-гистологическую целостность мозга, в то время как в мозжечке и спинном мозгу встречается множество поражений. В мозжечке обнаруживается атрофия коры, не затрагивающая узелок и вентральный клочок, как и околоклочковую зону мозжечка, крышечные ядра и миндалины. Атрофия нервных клеток и разрежение миелиновых волокон, встречающиеся постоянно, объясняют умственную отсталость, представляемый этими больными. В спинном мозгу определяется утолщение пирамидальных пучков. Течение синдрома Маринеску–Шегрена. Мозжечковые нарушения прогрессивно усиливаются и, одновременно, психический дефицит. Лечение синдрома Маринеску–Шегрена. Не существует эффективного лечения в большинстве случаях, вследствие чего предписываются некоторые меры профилактического характера: избежание единокровия у родителей; генеалогическое исследование больных. Генетический совет особенно полезен в жизни больных этим синдромом; долг врача осведомить семью о наследственном характере аномалии и привлечь внимание на возможность ее появления у наследников. Родители, осведомленные о положении, могут решить иметь ли им детей или нет. Скопировано с сайта: http://www.detvrach.com - Детский врач
Сцепленная с полом глухота
MB Petersen, Q Wang, PJ Willems
Clinical Genetics Volume 73 Issue 1 Page 14-23, January 2008 doi:10.1111/j.1399-0004.2007.00913.x
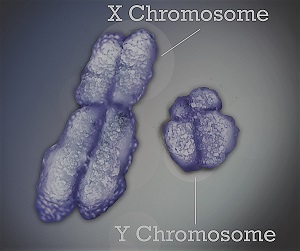 Многие генетические синдромы характеризуются глухотой (1). Также несиндромальная потеря слуха встречается часто: 1 на каждые 2000 новорожденных имеет моногенную форму несиндромальной доречевой глухоты (2, 3), а многие обнаруживают послеречевую потерю слуха. Сегодня более 100 локусов участвуют в несиндромальной потере слуха и 40 ядерных генов, вызывающие несиндромальную потерю слуха, идентифицированы в последние 15 лет (4-17). Все эти локусы и соотв. гены представлены online (Hereditary Hearing Loss Homepage of the University of Antwerp). Спектр генов включает цитоскелетные белки, такие как миозины, структурные белки, такие как альфа текторин и коллаген 11A2, транскрипционные факторы, такие как POU3F4 и белки ионных каналов, такие как KCNQ4 и коннексины.
Многие генетические синдромы характеризуются глухотой (1). Также несиндромальная потеря слуха встречается часто: 1 на каждые 2000 новорожденных имеет моногенную форму несиндромальной доречевой глухоты (2, 3), а многие обнаруживают послеречевую потерю слуха. Сегодня более 100 локусов участвуют в несиндромальной потере слуха и 40 ядерных генов, вызывающие несиндромальную потерю слуха, идентифицированы в последние 15 лет (4-17). Все эти локусы и соотв. гены представлены online (Hereditary Hearing Loss Homepage of the University of Antwerp). Спектр генов включает цитоскелетные белки, такие как миозины, структурные белки, такие как альфа текторин и коллаген 11A2, транскрипционные факторы, такие как POU3F4 и белки ионных каналов, такие как KCNQ4 и коннексины.
Первый ядерный ген, участвующий в несиндромальной потере слуха идентифицирован в 1995; он оказался X-сцепленным POU3F4 геном (18). Хотя впечатляющее количество ядерных генов было позднее установлено при несиндромально потере слуха, второй Х-сцепленный ген не был идентифицирован.
Опубликованы многочисленные обзоры по аутосомной и митохондриальной потере слуха (4-17), но отсутствуют публикации о сцепленой с полом потере слуха. Это исследование дополняет наши три предыдущие обзора по аутосомно доминантной (4), аутосомно рецессивной (5) и митохондриальной (6) несиндромальной потере слуха.