Одним из ключевых параметров лигандов TSPO, определяющих их биологическую активность, является их аффинность по отношению к этому рецептору. В связи с этим нами был проведен анализ теоретической аффинности новых лигандов TSPO в ряду 1-арилпирроло[1,2- a ]пиразин-3-карбоксиамидов с использованием QSAR и метода молекулярного докинга.
QSAR/QSPR – метод измерения количественной связи между структурой молекулы и её свойством, в нашем случае -активностью. Методы QSAR появились в начале 1960-х и их основателем был Корвин Ганч. Основное применение QSAR моделей- прогноз биологической активности для химических соединений[50]. Для числового обозначения молекулы используют независимые друг от друга числовые параметры молекулы - дескрипторы. [51]
Существующие QSAR методы можно разбить на две группы: на основе трехмерной структуры молекулы и на основе топологии молекулы, которые используют информацию о атомах и типах связи между ними, а также другие локальные дескрипторы (липофильность, константы заместителей, дипольный момент и др.). Структурная формула молекул записывается в виде молекулярного графа, где атомы — это вершины, а ребра – это связи между атомами. Однако топологический подход не позволяет учитывать информацию о взаимном расположении структурных особенностей в пространстве и такие методы не могут быть универсальным инструментом прогнозирования физиологической активности.
Для построения QSAR моделей формируется обучающая и тестовая выборка из молекул для которых заранее известна биологическая активность. Обучающая выборка загружается в компьютерную программу, которая проводит подбор дескрипторов и строит зависимость как А = f(D), где A- активность, D – дескриптор. На основе обучающей выборки строится корреляционное уравнение активности из набора дескрипторов. Модель проверяется с помощью тестовой выборки, в которой активность молекул уже известна. Таким образом модель связывает активность с определенными фрагментами в молекуле и их окружением. Точность модели зависит от размера обучающей выборки и оптимального набора дексрипторов. [52]
Анализ связи структура-активность [24] (Таблица 4) показал, что наибольшую активность проявляют соединения, у которых отсутствует заместитель в бензольном кольце (R3 = H), а амидная группа замещена на метильную (R1= CH3) и бензильную или н-бутильную группу (R2= Bz, n-Bu). Различные вариации орто заместителя R3 в бензольном кольце (ГМЛ-2, ГМЛ-4, ГМЛ-5) при R1=CH3, R2=n-Bu, приводят к снижению активности. При введении фтора в R3 положение активность снижается, а при введении брома и хлора полностью исчезает. Удаление одного из заместителей у амидной группы (ГМЛ-7 и ГМЛ-8) R1 или R2 = H существенно снижает активность.
Таблица 4.

| Шифр
| R1
| R2
| R3
|
| ГМЛ – 1
| CH3
| Bz
| H
|
| ГМЛ – 2
| CH3
| n-Bu
| Cl
|
| ГМЛ – 3
| CH3
| n-Bu
| H
|
| ГМЛ – 4
| CH3
| n-Bu
| F
|
| ГМЛ – 5
| CH3
| n-Bu
| Br
|
| ГМЛ – 6
| CH3
| втор-Bu
| H
|
| ГМЛ – 7
| H
| Bz
| H
|
| ГМЛ – 8
| H
| CH3
| H
|
| ГМЛ – 9
| CH3
| Bz
| F
|
| ГМЛ – 10
| CH3
| Bz
| Cl
|
| ГМЛ – 11
| CH3
| изо-Bu
| H
|
Для анализа количественной связи структура-активность построена QSAR модель на основе обучающей выборки из 1297 соединений с известной константой ингибирования. Первоначально осуществили поиск лигандов TSPO с известными константами ингибирования в базе данных химических структур ZINC [53]. Полученную базу в формате sdf загрузили в веб-сервис для QSAR моделирования - OCHEM [54]. Были созданы 9 моделей из комбинации трех методов машинного обучения (ASNN, FSMLR, PLS) с различным набором 2D фрагментарных дескрипторов (OEState, ISIDA, GSFrag). Для валидации модели использовался 5 – кратный перекрёстный контроль. В результате была выбрана модель на основе искусственных нейронных сетей с коэффициентом корреляции r2= 0,68 (Таблица 5) и параметром перекрёстного контроля q2= 0,65 (Таблица 6). Графическое отображение модели изображено на рисунке 5. В качестве дескрипторов были использованы фрагментарные дескрипторы ISIDA длинной 2-4 атома.
Таблица 5. Значения коэффициента корреляции для комбинаций дескрипторов и методов машинного обучения.
| R2
| ASNN
| FSMLR
| PLS
|
| ALogPS, OEstate
| 0.62
| 0.32
| 0.41
|
| Fragmentor (Length 2 - 4)
| 0.68
| 0.41
| 0.5
|
| GSFrag
| 0.67
| 0.22
| 0.33
|
Таблица 6. Значения коэффициента перекрестного контроля для комбинаций дескрипторов и методов машинного обучения.
| Q2
| ASNN
| FSMLR
| PLS
|
| ALogPS, OEstate
| 0.6
| 0.24
| 0.39
|
| Fragmentor (Length 2 - 4)
| 0.65
| 0.4
| 0.48
|
| GSFrag
| 0.64
|
| 0.32
|

Рис.5 Графическое отображение прогностической модели от фрагментарных дескрипторов ISIDA
Фрагментарные дескрипторы ISIDA базируются на двух классах дескрипторов: подструктурные молекулярные фрагменты и фармакофорные триплеты. Подструктурные молекулярные фрагменты разбиты на три класса: (1) последовательность соединенных атомов и связей, (2) только атомы или пары атомов, (3) расширенный набор атомов с близкой средой и их связей. Фармакофорные триплеты производят поиск и выделение определенных фармакофорных фрагментов (H-доноры, H-акцепторы, анионы, катионы и т.д) на заданных топологических расстояниях [55].
В результаты были получены прогнозируемые значения аффинности для 22 структур (Таблица 7) с различными заместителями у амидной группы.
Таблица 7. Значения прогнозируемой активности от фрагментарных дескрипторов ISIDA.
| №
| R1
| R2
| Ki, -log(M)
| №
| R1
| R2
| Ki, -log(M)
|
|
| Me
| H
| 5,3
|
| Ph
| n-Bu
| 6,2
|
|
| Et
| Et
| 5,5
|
| Bz
| Me
| 6,9
|
|
| Pr
| Pr
|
|
| Bz
| Et
| 5,7
|
|
| Me
| n-Bu
|
|
| Bz
| n-Bu
| 6,2
|
|
| Me
| i-Bu
|
|
| Bz
| i-Pr
| 5,4
|
|
| Me
| sec-Bu
| 6,4
|
| Bz
| Bz
| 5,5
|
|
| цикло
| гексил
| 5,1
|
| Ph
| Bz
| 5,8
|
|
| Bz
| H
| 5,2
|
| Ph
| Ph
| 6,1
|
|
| Ph
| Et
| 5,7
|
| Ph
| циклогексил
| 5,2
|
|
| Ph
| Pr
| 6,1
|
| Ph
| циклопентил
| 5,6
|
|
| Ph
| Me
| 6,4
|
| Naph
| H
| 5,6
|
Молекулярный докинг.
Метод молекулярного докинга удобный инструмент для расчета энергии связи «лиганд-белок» и поиска конформации лиганда связанного с белком. Программа осуществляет поиск глобального минимума, используя генетический алгоритм. В процессе докинга лиганд помещается в пространство, для которого заранее подготовлены электростатические карты для каждого химического элемента лиганда. В процессе моделирования генерируется начальная популяция лигандов порядка 300, затем рассчитывается разница энергий связывания между начальным положением и «мутантным». Выбирается лучшая конформация и процесс повторяется. В результате докинга мы получаем значения ∆G для комплекса лиганд-белок и положение молекулы соответствующее этой энергии. (Рис.3)
Расчет производится при помощи программного обеспечения AutoDock 4.2 [56]. Для оценки аффинности соединений использовалась структура TSPO млекопитающего (мыши) в комплексе с лигандом PK11195, полученная в высоком разрешении методом ЯМР‑спектроскопии [57].
Все исследованные лиганды были переведены в 3D структуры в программе Marvin Sketch, после чего использовались для in silico лиганд-белкового докинга. Молекулярный докинг производился в несколько этапов. Сначала из комплекса 2MGY был удален исходный лиганд PK11195, после чего в программе AutoDock к молекуле белка были добавлены водороды и посчитан заряд по методу Гейстегера[58]. Далее, 3D результат докинга визуализировался в программе PyMOL с обозначением поверхности кармана связывания (Рис.6)
Рис. 6 Трёхмерная модель активного центра TSPO.
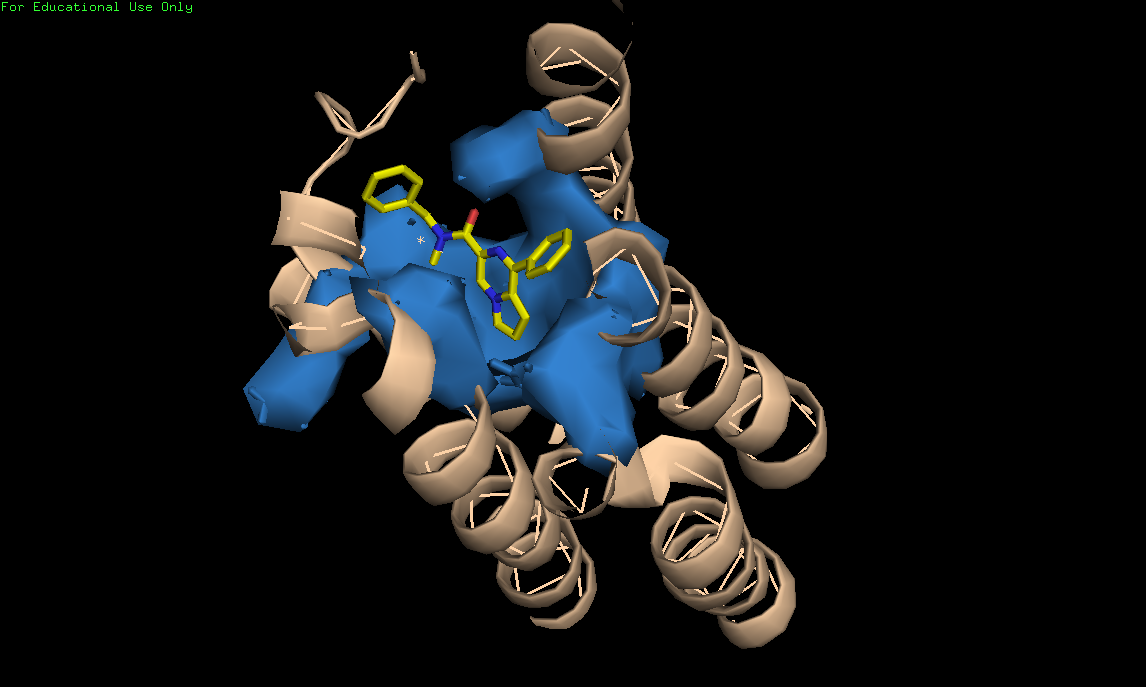
Таблица 8. Результаты молекулярного докинга при помощи программы AutoDock 4.2 
| №
| R1
| R2
| Ki, nM
| Clog P
| №
| R1
| R2
| Ki, nM
| Clog P
|
|
| Me
| H
|
| 1,89
|
| Ph
| n-Bu
|
| 5,09
|
|
| Et
| Et
|
| 2,82
|
| Bz
| Me
|
| 3,83
|
|
| Pr
| Pr
|
| 3,87
|
| Bz
| Et
|
| 4,19
|
|
| Me
| n-Bu
|
| 3,43
|
| Bz
| n-Bu
|
| 5,16
|
|
| Me
| i-Bu
|
| 3,35
|
| Bz
| i-Pr
|
| 4,61
|
|
| Me
| sec-Bu
|
| 3,4
|
| Bz
| Bz
|
| 5,56
|
|
| цикло
| гексил
|
| 2,96
|
| Ph
| Bz
|
| 5,49
|
|
| Bz
| H
|
| 3,61
|
| Ph
| Ph
|
| 5,42
|
|
| Ph
| Me
|
| 3,77
|
| Naph
| H
|
| 4,89
|
|
| Ph
| Et
|
| 4,12
|
| Ph
| циклопентил
|
| 5,48
|
|
| Ph
| Pr
|
| 4,65
|
| Ph
| циклогексил
|
| 5,57
|
Дальнейшему анализу подвергли самые аффинные соединения (17,18,19,20) и соединения с Clog P <4,5 (9, 10, 13, 14). В результате докинга определилось два основных мотива связывания.
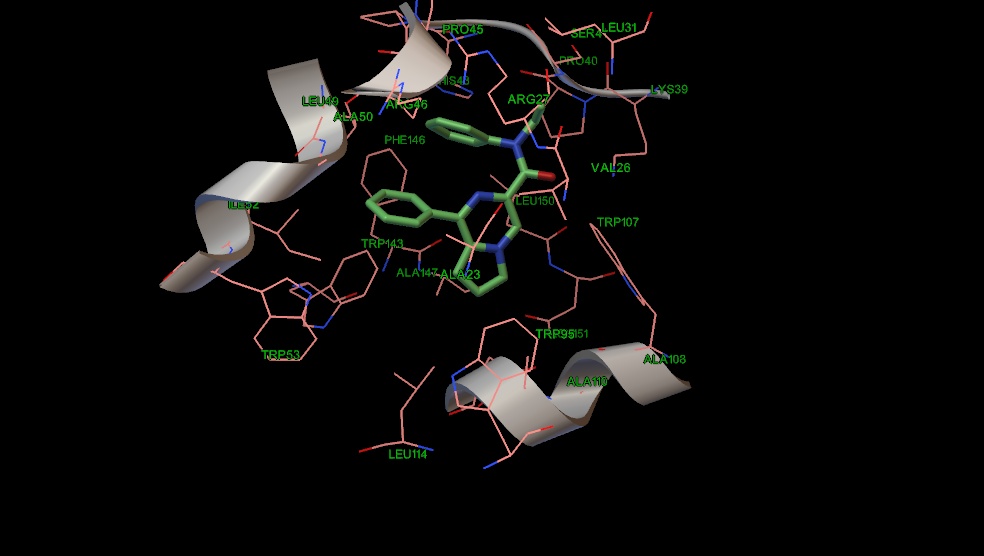
В одном мотиве пирролопиразиновый остов наиболее аффинных соединений 18 и 19 гидрофобно взаимодействует с ароматическими остатками триптофана (95,107), аланина (110, 147) и лейцина 150, а липофильные заместители амидной группы с лейцином 150, серином 41, триптофаном 143, изолейцином 52. Фенильное кольцо у гетероароматического остова располагается около двух остатков триптофана 53 и 95 (Рис 7,8). Соединение 10 имеет похожий мотив связывания, однако фенильное кольцо в гетероцикле несколько сдвинуто к остатку триптофана 143 (Рис. 9), в отличие от соединений 18 и 19, которые не могут сдвинуться из-за наличия объемных заместителей у амидной группы.
Рис.7 Положение соединения 18 в сайте связывания PK-11195.

Рис. 8 Положение соединения 19 в сайте связывания PK-11195.

Рис. 9 Положение соединения 10 в сайте связывания PK-11195.
Альтернативный мотив связывания предсказан для структур (9, 13, 14, 17,20). В отличие от соединений 10,18,19 гетероциклический остов молекул 13 и 17 (Рис 10,11) взаимодействует с остатками триптофана (53,143), изолейцина 52 и аланина 23. Однако у соединений 9, 14, 20 фенильное кольцо сдвигается в сторону стэкинг взаимодействия с триптофаном 107, что в итоге сдвигает пирролопиразиновый цикл к остатку триптофана 95 и лейцина 114 (Рис 12,13,14). У амидного атома азота соединений 13 и 17 положения бензильной группы совпадают, а насыщенные атомы углерода направлены к лейцину 49. Этильный радикал у амидного азота соединения 14 также направлен к лейцину 49, а бензильная группа к валину 26. Соединение 9 тоже несколько сдвинуто по метильному радикалу к остатку лейцина 49. Нафтильная группа в соединении 20 имеет дополнительную область связывания с валином 26, лейцином 31, серином 41 и аргинином 46. Таким образом, введение электроноакцепторной группы в параположение фенильного кольца у атома амидного азота может увеличить сродство к TSPO.
Рис. 10 Положение соединения 13 в сайте связывания PK-11195.
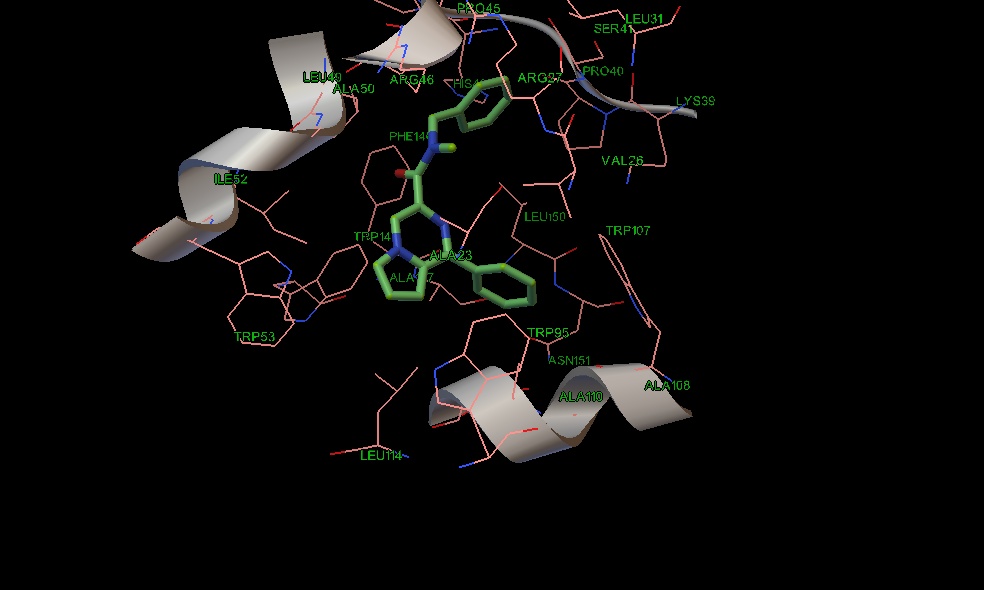
Рис. 11 Положение соединения 17 в сайте связывания PK-11195.

Рис. 12 Положение соединения 9 в сайте связывания PK-11195.
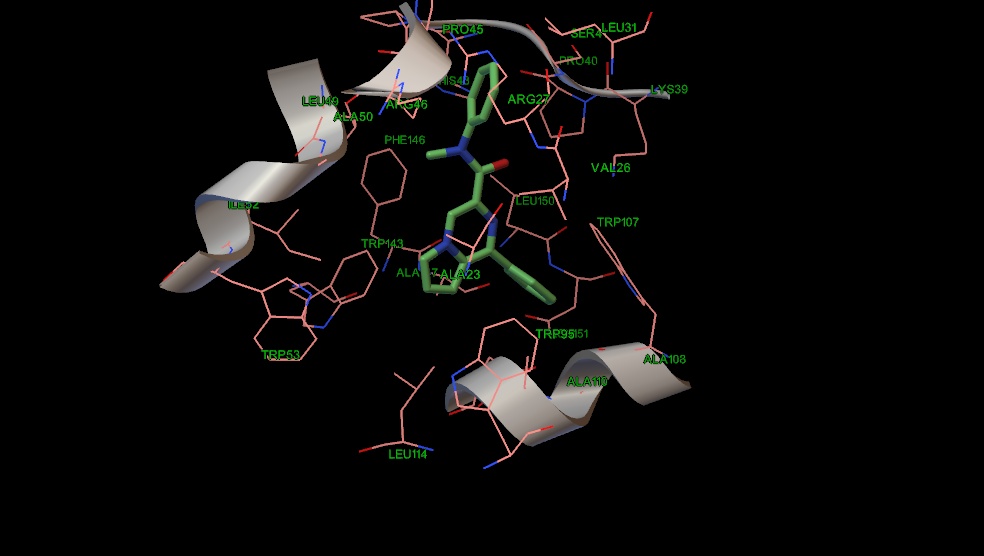
Рис. 13 Положение соединения 14 в сайте связывания PK-11195.

Рис. 14 Положение соединения 21 в сайте связывания PK-11195.
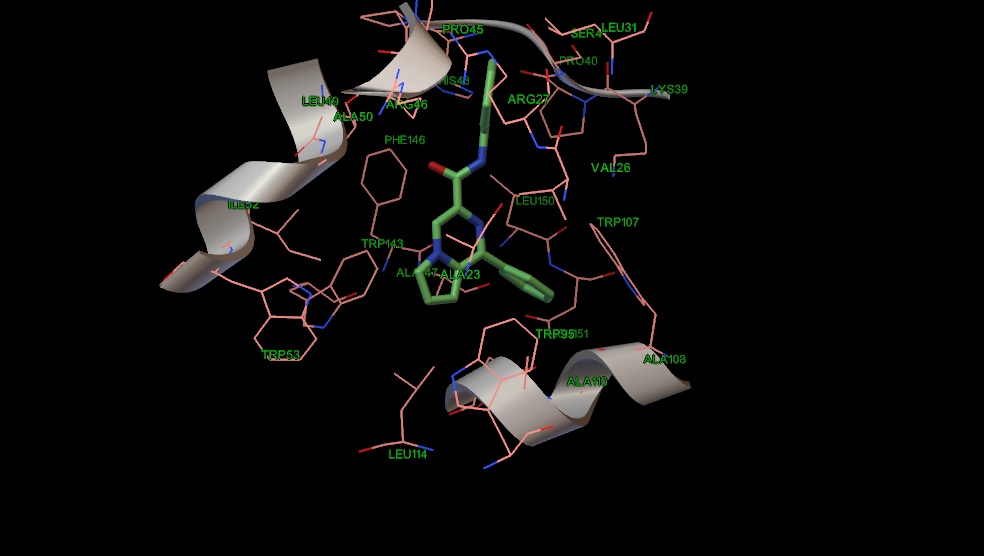
По причине того, что карман активного центра PK-11195 достаточно гидрофобен, проведен анализ соответствия гидрофобной поверхности лиганда к поверхности белка с помощью веб-сервиса PLATINUM. В этом веб-сервисе производятся вычисления, основанные на концепции молекулярного гидрофобного потенциала, который присваивается каждому атому в зависимости от его природы. Затем производится анализ поверхности молекулы лиганда и его окрестностей. Результат -это число, полученное при делении гидрофобно комплементарной поверхности лиганда к белку на общую поверхность лиганда. Также на основе эмпирических функций ведется учет стэкинговых взаимодействий между ароматическими остатками аминокислот и ароматическими фрагментами лигандов. Результаты исследуемых структур сравнивали с результатами классического лиганда TSPO PK-11195 (Таблица 9).
Таблица 9. Результаты проверки гидрофобного соответствия в веб-сервисе PLATINUM.
| Номер молекулы
| π – π стэкинг
| Соответствие
|
|
| 1,65
| 0,8859
|
|
| 0,87
| 0,9459
|
|
| 2,42
| 0,9139
|
|
| 2,03
| 0,9371
|
|
| 1,78
| 0,8971
|
|
| 0,71
| 0,9235
|
|
| 0,95
| 0,9339
|
|
| 1,76
| 0,8701
|
| PK-11195
| 1,97
| 0,9609
|
В итоге вышеперечисленных прогнозов, принимая во внимание коэффициент clog P и результаты соответствия гидрофобных поверхностей, соединения 13,16,18,19,20 не подходят к правилу Липински clog P < 5[59]. Оптимальные значения log P для проникновения через гематоэнцефалический барьер лежат в интервале 1.5-2.7[60]. Таким образом, перспективными соединениями, учитывая липофильность, аффинность и гидрофобный потенциал, стали соединения 9, 10, 13, 14.
В результате исследований теоретических расчетов липофильности и аффинности к TSPO пирроло[1,2-a]пиразинов был сделан вывод, что N-метил-N-фенил-1-фенилпирроло[1,2-a]пиразин-3-карбоксамид(9) (Рис.15) и N-этил-N-фенил-1-фенилпирроло[1,2-a]пиразин-3-карбоксамид (10) (Рис.16) подходят под фармакофорную модель и могут потенциально являться инновационными анксиолитиками.


Рис.15 Рис.16
Синтез
Исходя из доступных реагентов и методик синтеза фенил замещенных пирролпиразинов, был выбран метод получения гетероароматического цикла из реакции пирролфенилкетона с 2-азидоакриламидами. Нами предложен план синтеза (Схема 3.1).
Схема 3.1

Альтернативный способ получения 1-фенилпирроло[1,2-a]пиразин-3-карбоксамидов заключается в получении 1-фенилпиролло[1,2-a]пиразин-3-карбоновой кислоты и последующем синтезе различных амидов (Схема 3.2)
Схема 3.2