Для того чтобы получше понять происхождение CoV2, давайте ещё раз взглянем на последовательности шиповидного белка у нашей троицы: CoV2, RaTG13 и панголина — сравним попарную разницу между ними (точками отмечены идентичные аминокислоты, красные буквы показывают разницу, а прочерки обозначают удалённые/добавленные аминокислоты):

Невооружённым глазом видно, что в первой четверти сиквенса панголиний штамм далёк от CoV2 и RaTG13. Ну а RaTG13, если бы не участок в районе RBM (красный прямоугольник), был бы ооочень близок к CoV2. Но, как я уже говорил, тот самый участок у CoV2 ближе всего к панголиньему штамму.
Кстати, а что там у других панголиньих штаммов? Давайте посмотрим. Ведь пока что мы анализировали только вирус, выделенный из панголинов, конфискованных таможней в 2019 году. А ещё была партия панголинов, конфискованных в 2017-ом, и у них тоже был выделен похожий штамм. Если сравнить RaTG13 с геномами вирусов из панголинов 2017 и 2019 годов, то тут тоже всё интересно:

В первой четверти S белка панголиньи штаммы от 2017 года ближе к RaTG13 (и CoV2) чем их панголиний собрат от 2019-го (MP789). При этом, у всех трёх явный недавний общий предок в областях, выделенных зелёными прямоугольниками, и в этих областях RaTG13 и панголин-19 (MP789) к нему ближе, чем панголин-17, так как у него с RaTG13 несколько общих мутаций (обведены красными и синими эллипсами), которых не наблюдается у легенды-17. При этом RBM у всех трёх разный, и разный примерно в одинаковой пропорции, и в схожих местах.
Возможно, уже после того как предки RaTG13 и MP789 разошлись, у MP789 произошла замена большого участка в первой четверти S белка (которой не произошло у RaTG13 и панголина-17), а остальная часть S белка у всех троих осталась общей. А потом пути генофондов RaTG13 и MP789 сошлись опять и в порыве страсти породили CoV2. Также вполне возможно, что предок RaTG13 является результатом рекомбинации предков панголиньих штаммов.
Ещё любопытно видеть довольно уникальную одинаковую мутацию (QTQTNS) у RaTG13 и панголина-19 прямо перед тем местом, где у CoV2 находится новый furin cleavage site, возникший из-за врезки 4-х новых аминокислот в этом месте (PRRA). Если посмотреть на нуклеотидную последовательность вокруг этой одинаковой мутации, то видно, что RaTG13 и CoV2 по ней ближе друг к другу, чем к панголину-19, так как успели накопить несколько общих мутаций (выделены синим):

Кстати, с Orf1ab у CoV2 тоже филогенетическая чехарда: 1a ближе к RaTG13, а вот 1b — к панголину-19:
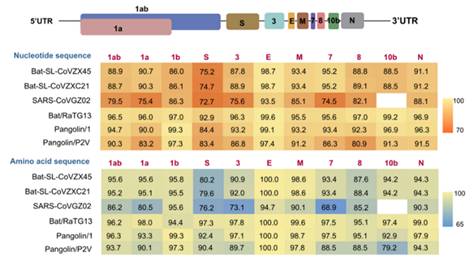
То есть получается, что предок CoV2 согрешил с общим предком панголина-19, как минимум, дважды? Впервые — когда он (вместе с общим предком с RaTG13) унаследовал Orf1ab и вторую часть шиповидного белка с QTQTNS мутацией, а вторично — когда приобрёл 1b вместе с RBM, уже отличающимся от RaTG13-шных. Нет, конечно, такое возможно и само по себе не особо примечательно — ведь эти вирусы мутируют и рекомбинируют постоянно. Другой вопрос, где именно летучемышиные и панголиньи вирусы могут друг с другом встречаться для таких оргий — в горных пещерах, “мокрых рынках”, приютах для конфискованных животных, или даже в лабораториях. Но давайте повременим с этими вопросами. Сначала рассмотрим самый бросающийся в глаза аспект нового вируса — врезку из 4-х аминокислот, превратившую его в супер-убийцу.
Врежу аккуратно, но сильно
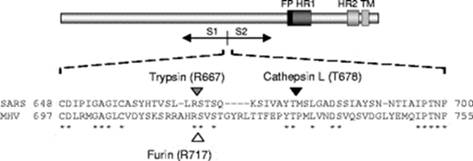
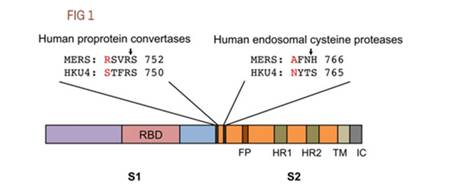
Совершенно невозможно игнорировать эту врезку PRRA между S1 и S2. Как заноза торчит она в геноме нашего нового CoV2. Врезка эта не простая, а золотая. Она создаёт тот самый furin cleavage site, о котором я упомянул в самом начале. Что это за зверь? Попытаюсь вкратце объяснить. Помните структуру нашего шиповидного белка? Вот наглядная диаграмма:
Белок состоит из двух частей, S1 и S2, из которых S1 отвечает за первичный контакт с рецептором (тот самый Receptor Binding Domain/Motif), а S2 отвечает уже за слияние с клеточной мембраной и проникновение в клетку. Запускает процесс слияния отмеченный жёлтым fusion peptide, но для того чтобы он начал своё грязное дело, кто-то должен разрезать S белок в одном из сайтов, выделенных ромбиками на диаграмме выше. Своих таких “резальщиков” у вируса нет (есть другие, но это к делу не относится), поэтому он полагается на различные протеазы своих жертв, благо таких протеаз, как можно понять по обилию цветов тех самых ромбиков, существует несколько видов. Но не все они равны, и не во всех типах клеток есть нужные вирусу протеазы. А фурин как раз один из самых эффективных, да ещё и обитает он не только на поверхности клеток, но и внутри. Нагляднее всего опасность нового фуринового сайта демонстрирует разница между CoV2 и его “предтечей” SARS-CoV:
Как видно из диаграммы, в случае CoV2, благодаря фуриновому сайту, разрезать его S белок вне клетки могут не два, а три класса протеаз (три разноцветных пакмэна). Но, быть может, самая важная разница состоит в том, что фурин присутствует и внутри клетки, поэтому он может разрезать S белок сразу после сборки вириона, тем самым повышая способность новых вирионов сливаться с другими клетками — так сказать, не отходя от кассы.
Кстати, существует вероятность, что именно новый фуриновый сайт играет важную роль в выраженной возрастозависимой морбидности и летальности CoV2:
По неизвестным причинам, пациенты с гипертонией, диабетом, ишемической болезнью сердца, цереброваскулярными заболеваниями, хронической обструктивной болезнью легких и почечной дисфункцией имеют худшие клинические исходы при инфицировании SARS-CoV-2. Целью данного обзора является обобщение доказательств существования повышенного плазмин(оген)а у пациентов с COVID-19 с этими коморбидными состояниями. Плазмин и другие протеазы могут расщеплять новый фуриновый сайт в шиповидном белке SARS-CoV-2 внеклеточно, что увеличивает его инфекционность и вирулентность.
Исходный текст
Patients with hypertension, diabetes, coronary heart disease, cerebrovascular illness, chronic obstructive pulmonary disease, and kidney dysfunction have worse clinical outcomes when infected with SARS-CoV-2, for unknown reasons. The purpose of this review is to summarize the evidence for the existence of elevated plasmin(ogen) in COVID-19 patients with these comorbid conditions. Plasmin, and other proteases, may cleave a newly inserted furin site in the S protein of SARS-CoV-2, extracellularly, which increases its infectivity and virulence.
Ах, да, разрезает фурин белки в строго определённых местах, а именно после последовательности RxxR (то есть Arg-X-X-Arg, где X может быть любой аминокислотой). При этом если на втором или третьем месте тоже аргинин (то есть RRxR или RxRR), то эффективность расщепления этого сайта существенно возрастает.
Поэтому появление нового furin cleavage site специалистами было замечено сразу. Ещё бы! Ведь такого сайта нет ни у кого из ближайших или даже дальних родственников Cov2 — те коронавирусы, у которых он есть, лишь на 40% близки к Cov2 по геному:
Исходный текст
It was found that all Spike with a SARS-CoV-2 Spike sequence homology greater than 40% did not have a furin cleavage site (Figure 1, Table 1), including Bat-CoV RaTG13 and SARS-CoV (with sequence identity as 97.4% and 78.6%, respectively). The furin cleavage site “RRAR” in SARS-CoV-2 is unique in its family, rendering by its unique insert of “PRRA”. The furin cleavage site of SARS-CoV-2 is unlikely to have evolved from MERS, HCoV-HKU1, and so on. From the currently available sequences in databases, it is difficult for us to find the source. Perhaps there are still many evolutionary intermediate sequences waiting to be discovered.
Вот наглядная иллюстрация из той же статьи, что и вышеприведённая цитата (розовым отмечены коронавирусы с фуриновым сайтом, на 10 часах приведены 3 разных штамма Cov2):
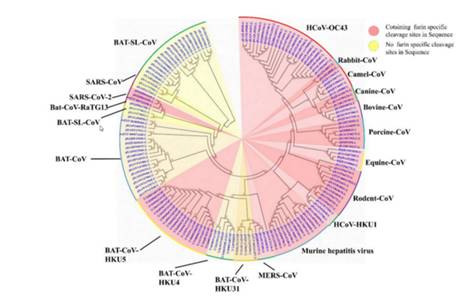
Ближайший родственник с фуриновым сайтом — это штамм HKU5, выделенный командой Ши Чжэнли в 2014 году в Гуанчжоу из летучих мышей рода Pipistrellus (добавлен в GenBank в 2018-ом). Но родственник он весьма дальний — их шиповидные белки совпадают лишь на 36%.
В общем, учёные в замешательстве. Откуда взялась эта вставка из 12 нуклеотидов? Может ли она быть рукотворной? Вполне. Ведь такими вставками вирусологи занимались многократно, причём издавна. Например, американцы вставляли RRSRR в шиповидный белок первого SARS-CoV ещё в 2006-ом:
Исходный текст
To investigate whether proteolytic cleavage at the basic amino acid residues, were it to occur, might facilitate cell–cell fusion activity, we mutated the wild-type SARS-CoV glycoprotein to construct a prototypic furin recognition site (RRSRR) at either position.

А японцы вставили такой сайт (RRKR) в белок SARS-CoV в 2008 году, правда немного ниже по течению:
В том же 2008-ом году их голландские коллеги тоже изучали эти протеазные сайты у SARS-CoV и сравнивали их с мышиным коронавирусом MHV, у которого этот сайт есть (SRRAHR|SV), причём похожий на сайт нашего CoV2 (SPRRAR|SV):
В 2009 году другая американская группа тоже решила поупражняться над “улучшением” SARS-CoV и, не изменяя американской традиции не мелочиться с аргининами, они вставили RRSRR:
Исходный текст
To examine the potential use of the SARS-CoV S1–S2 and S2? positions as sites for proteolytic cleavage, we first introduced furin cleavage recognition sites at these locations by making the following mutations 664-SLLRSTSQSI — SLLRRSRRSI-671 (S1–S2) and 792-LKPTKRSF — LKRTKRSF-799 (S2?).
Пекин-2019
Но самой недавней подобной работой, которую я видел, была работа октября 2019 года учёных из Пекина, где новый фуриновый сайт RRKR вставили не в какой-то псевдовирус, а в самый настоящий куриный коронавирус, infectious bronchitis virus (IBV):
Кстати, интересно упоминание авторов, что добавление фуринового сайта позволяет вирусу-мутанту поражать нервные клетки. Быть может, именно фуриновый сайт CoV2 является причиной того, что некоторые пациенты с CoV2 демонстрируют неврологическую симптоматику, включая потерю обоняния:
Мутация сайта S2' шиповидного белка рекомбинантного вируса генотипа QX приводит к более высокой патогенности, выраженным нервным симптомам и нейротропизму по сравнению с цыплятами, инфицированными вурусом дикого типа IBV (WT-IBV). В этом исследовании мы представляем доказательства того, что рекомбинантный IBV с мутантным сайтом S2 (фуриновый сайт-S2) приводит к более высокой смертности. Заражение мутантом IBV вызывает тяжелый энцефалит и разрушает гематоэнцефалический барьер.
…
Таким образом, наши результаты демонстрируют, что фуриновый сайт перед FP в шиповидном белке является важным сайтом для CoV, модулирующим проникновение, слияние клеток с вирусом, адаптацию к клетке-хозяину, клеточный тропизм и патогенность, но не антигенность.
Исходный текст
Mutation of the S2' site of QX genotype (QX-type) spike protein (S) in a recombinant virus background results in higher pathogenicity, pronounced neural symptoms and neurotropism when compared with conditions in wild-type IBV (WT-IBV) infected chickens. In this study, we present evidence suggesting that recombinant IBV with a mutant S2' site (furin-S2' site) leads to higher mortality. Infection with mutant IBV induces severe encephalitis and breaks the blood–brain barrier.
…
In summary, our results demonstrate that the furin cleavage site upstream of the FP in S protein is an important site for CoV, modulating entry, cell–virus fusion, adaptation to its host cell, cell tropism and pathogenicity, but not antigenicity.
При этом у многих коронавирусов фуриновые сайты, конечно же, встречаются в природе, и они весьма разнообразны. Да и появляться они могут и в результате случайных мутаций. Именно это произошло в случае с MERS-ом, о чем в 2015 нам поведал международный коллектив авторов, среди которых были и Ши Чжэнли, и Ральф Барик — две звезды синтетической коронавирусологии. О них мы ещё неоднократно вспомним, а пока пару слов о той статье. Собственно в ней авторы показали две ключевые мутации, которые позволили MERS перескочить с летучих мышей на человека. И одна из этих мутаций привела к возникновению фуринового сайта. Правда это была не врезка новых аминокислот, а мутации ранее существующих (отмеченных красным ниже):
Авторы не просто показали, а привязали эти мутации обратно к исходному летучемышиному шиповидному белку, создав в нём те же самые мутации (то есть, и фуриновый сайт), и показав, что это придаёт ему способность заражать человеческие клетки:
Исходный текст
To evaluate the potential genetic changes required for HKU4 to infect human cells, we reengineered HKU4 spike, aiming to build its capacity to mediate viral entry into human cells. To this end, we introduced two single mutations, S746R and N762A, into HKU4 spike. The S746R mutation was expected to restore the hPPC motif in HKU4 spike, whereas the N762A mutation likely disrupted the potential N-linked glycosylation site in the hECP motif in HKU4 spike.
…
Therefore, the reengineered hPPC and hECP motifs enabled HKU4 spike to be activated by human endogenous proteases and thereby allowed HKU4 pseudoviruses to bypass the need for exogenous proteases to enter human cells. These results reveal that HKU4 spike needs only two single mutations at the S1/S2 boundary to gain the full capacity to mediate viral entry into human cells.
Кстати, то как они это сделали может людей далёких от современных биотехнологий напугать само по себе — потому что авторы вставили этот коронавирусный шиповидный белок в инактивированный ретровирус (ВИЧ):
Вкратце, псевдотипированные ретровирусы MERS-CoV-spike, экспрессирующие репортерный ген люциферазы, были получены путем котрансфекции клеток HEK293T плазмидой, несущей геном ВИЧ-1, дефектный по Env, экспрессирующий люциферазу (pNL4–3.luc.RE-) и плазмидой, кодирующей MERS-CoV шиповидный белок.
Исходный текст
Briefly, MERS-CoV-spike-pseudotyped retroviruses expressing a luciferase reporter gene were prepared by cotransfecting HEK293T cells with a plasmid carrying Env-defective, luciferase-expressing HIV-1 genome (pNL4–3.luc.R-E-) and a plasmid encoding MERS-CoV spike protein.
Быть может, именно это подвигло индийских исследователей на поиск схожих у ВИЧ и CoV2 последовательностей в геноме (но их препринт быстро раскритиковали за неудачную методологию и ошибочные выводы). На самом деле такие псевдовирусы специалисты используют регулярно, и вообще, не стоит огульно бояться ретровирусов как класс — их подвид лентивирусы используется для генной терапии уже много лет.
Откуда взялся RaTG13
Вообще, RaTG13 — феноменальный штамм. Странно, что все эти годы группа Ши Чжэнли о нём молчала. Ведь он совсем не похож на остальных своих собратьев, особенно в последовательности шиповидного белка, а это, напомню, именно то место, которое определяет к каким типам клеток (и каких видов) данный вирус сможет цепляться. Вот график схожести генома CoV2 в сравнении с другими летучемышиными коронавирусами (панель B):
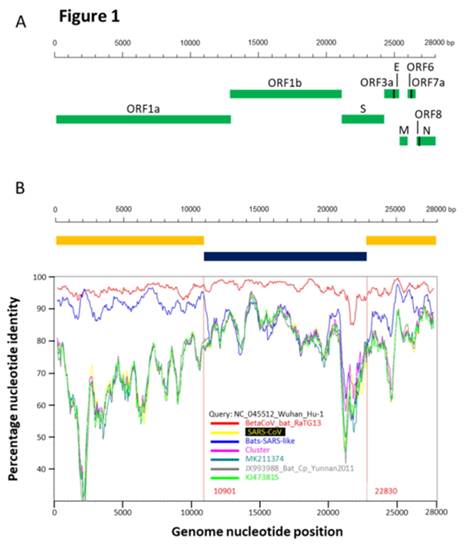
RaTG13 — это красная кривая, а синяя — это наиболее близкие к RaTG13 штаммы (ZXC21 и ZC45). Эти штаммы были выделены из Китайских подковоносов (Rhinolophus sinicus) в Чжоушане в 2015 (ZXC21) и 2017 (ZC45) годах. Как видно из графика, даже они очень сильно расходятся с RaTG13 (красная кривая) в районе S белка. При этом график выше не так хорошо передаёт масштаб пропасти между ними как прямое сравнение сиквенсов:

Как видим, шиповидные белки ZXC21 и ZC45 не только, в целом, на 23–24 аминокислотных остатка короче белка RaTG13, но они короче в самом важном месте — в RBM (см. делеции в красном прямоугольнике, отмеченные красными прочерками).
Так откуда же взялся RaTG13? Как я уже сказал, в 2020 году Ши Чжэнли сообщила, что выделила его у подковоносов (только вида Rhinolophus affinis, а не R. sinicus) в июле 2013 года в Юньнани. Правда, до конца января 2020 года публично о его существовании не сообщалось, а вот как описывает своё знаменательное открытие о его схожести с CoV2 сама группа Ши Чжэнли:
Затем мы обнаружили, что короткий участок РНК-зависимой РНК-полимеразы (RdRp) от коронавируса летучей мыши (BatCoV RaTG13), который ранее был обнаружен у Rhinolophus affinis из провинции Юньнань, показал высокую идентичность последовательности 2019-CoV2. Мы провели полноразмерное секвенирование на этом образце РНК (регистрационный номер GISAID EPI_ISL_402131). Анализ Simplot показал, что 2019-CoV2 очень похож на RaTG13 (Fig. 1c), с общей идентичностью генома 96,2%.
Исходный текст
We then found that a short region of RNA-dependent RNA polymerase (RdRp) from a bat coronavirus (BatCoV RaTG13) — which was previously detected in Rhinolophus affinis from Yunnan province — showed high sequence identity to 2019-CoV2. We carried out full-length sequencing on this RNA sample (GISAID accession number EPI_ISL_402131). Simplot analysis showed that 2019-CoV2 was highly similar throughout the genome to RaTG13 (Fig. 1c), with an overall genome sequence identity of 96.2%.
Негусто: ну был у них этот штамм «ранее обнаружен», и был. Лежал на полке. До 2020 года секвенировали в нём только часть генома, отвечающую за RdRp. Откуда он на полке взялся? В Юньнани в 2013 году выделили. Где именно? Не сказали. В GenBank тоже не было этой информации. Но, к счастью, в базе геномов GISAID была: сообщается, что в городе Пуэр (да, на родине любимого чая Басты) его выделили из fecal swab некого самца:

Это меня немного заинтриговало, потому что в моих блужданиях по Пабмеду, я уже натыкался на экспедицию в Пуэр летом 2013 года:
Исходный текст
Bats were captured from various locations in five counties of four prefectures of Yunnan Province, China, from May to July 2013.
В той экспедиции ничего особо интересного для нас исследователи не нашли, но, быть может, именно тогда Ши Чжэнли (или кто-то из её группы?) выделил тот самый образец RaTG13? Который они секвенировали лишь частично, но почему-то решили не публиковать, хотя он весьма отличался от всего известного ранее.
Сама Ши Чжэнли вполне могла лично участвовать в той экспедиции, потому что о таких экспедициях она отзывалась весьма тепло — например, в своём TED-like выступлении в 2018 году, где она показывала свои фотографии из таких экспедиций:
Именно серия таких экспедиций и принесла ей мировую славу и прозвище «Бэтвумен»: в 2013 году в статье в Nature группа Ши Чжэнли триумфально объявила о том, что в пещерах Юньнани она обнаружила летучих мышей-носителей штаммов RsSHC014 и Rs3367, которые совпадали с первым SARS-CoV на 85% и 96% соответственно.
Кстати, забавное совпадение, что примерно в то же время, в той же Юньнани группа Ши Чжэнли, оказывается, также обнаружила и штамм RaTG13, который оказался наиболее близок к CoV2, и их геномы тоже совпадают на 96%.
«Ухань-1»
Возвращаясь к той триумфальной статье от 2013 года, в ней же группа Ши Чжэнли ещё рассказала, что с помощью культивирования выделенных образцов в культуре обезьяньих клеток Vero, им удалось выделить живой вирус, который был практически идентичен штамму Rs3367 (наиболее близкому к SARS-CoV). Своё детище авторы окрестили WIV1 (где WIV обозначает Wuhan Institute of Virology):
Что наиболее важно, мы сообщаем о первой изоляции живого вируса SL-CoV (летучемышиный SL-CoV-WIV1) из образцов, выделенных из фекалий летучей мыши, в клетках Vero E6, который имеет типичную морфологию коронавируса, идентичность генома на 99,9% с Rs3367 и использует ACE2 людей, цивет и подковоносов для проникновения в клетку. Предварительное тестирование in vitro показывает, что WIV1 также имеет широкий видовой тропизм.
Исходный текст
Most importantly, we report the first recorded isolation of a live SL-CoV (bat SL-CoV-WIV1) from bat faecal samples in Vero E6 cells, which has typical coronavirus morphology, 99.9% sequence identity to Rs3367 and uses ACE2 from humans, civets and Chinese horseshoe bats for cell entry. Preliminary in vitro testing indicates that WIV1 also has a broad species tropism.
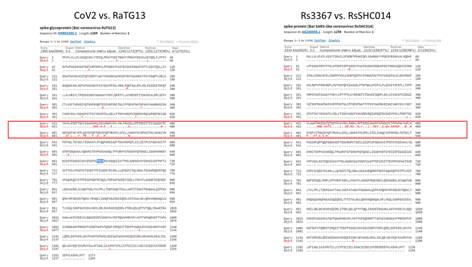
Давайте сравним RaTG13 со штаммами Rs3367 и RsSHC014:
Как видим, шиповидный белок этих штаммов не только короче RaTG13-шного на 13 аминокислот, но и сильно отличается он него в первой его четверти. Кстати, любопытно, что шиповидные белки у Rs3367 (aka WIV1) и RsSCH014 практически идентичны, и различаются только в районе RBD (правый сиквенс ниже). Почти как CoV2 и RaTG13 (не считая фуриновой врезки):
Мог ли какой-нибудь исследователь, получив в марте 2019-го образцы коронавируса из конфискованных таможней панголинов, захотеть проверить, насколько панголиний RBM тропен к человеческому рецептору? А если ещё и с polybasic фуриновым сайтом в самом интересном месте? Вот это будет бомба!
Теоретически, конечно, мог бы. Ничего технически сложного для вирусологов в осуществлении таких экспериментов нет. Резонный вопрос: а зачем им для этого использовать RaTG13 в качестве основы, а не уже опробованный WIV1? Но вполне возможно, что химеру с WIV1 они тоже тестировали. А параллельно решили смоделировать вариант рекомбинации панголиньего вируса с более близким ему летучемышиным — ведь RaTG13 всё же ближе к панголиньим штаммам чем WIV1: его шиповидный белок ближе к ним и филогенетически, и структурно — он с ними даже по длине совпадает, в то время как белки WIV1/Rs3367 и RsSHC014 на 13 аминокислот короче панголиньих. Да и общая для RaTG13 и панголина-19 (MP789) мутация QTQTNS в районе протеазного сайта не может оставить равнодушной специалиста.
Другие юньнаньские штаммы
Кстати, другие исследователи в 2011 году тоже находили образцы коронавирусов у Юньнаньских Rhinolophus affinis. Самым интересным мне показался штамм LYRa11:

Но и он весьма далёк от RaTG13, а куда ближе к Rs3367 (это тот штамм, что на 96% совпадает с первым SARS-CoV):

А вот RaTG13, выделенный из тех же летучих мышей Rhinolophus affinis, что и LYRa11, на него похож меньше всего (левый бласт).
Ну и наконец, ещё один Юньнаньский штамм (бесхитростно названный Yunnan2011), выделенный в 2011 году из другого подвида подковоносов, Rhinolophus pusillus, похож на RaTG13 ещё меньше чем LYRa11:

Да и между собой Yunnan2011 и LYRa11 (правый бласт выше) не особо похожи, не считая высококонсервативную область S2. Кстати, что за бардак у них с названиями штаммов? То полностью год прописывают, то частично, а то и вовсе нет (Rs3367). То сначала идёт вид носителя (Ra TG13), то потом (LY Ra 11). Ещё вот интересно, что обозначают TG, LY или SHC? Инициалы расшифровавшего геном?
Ладно, перейдём уже от вирусной археологии к вирусной инженерии, а именно к пересадке ключевых участков шиповидного белка и прочим gain-of-function (GOF) экспериментам.
Первый химерный коронавирус
Eсли вы думаете, что вся эта GOF-движуха с анализом того, что именно позволяет коронавирусам перепрыгивать с одного вида на другой началась в ответ на эпидемию первого SARS-а в 2002 году, вы ошибаетесь. Вирусологи экспериментировали с химерными коронавирусами задолго до этого. Вот, например, показательная статья от 1999 года от голландской группы Питера Роттиера из Утрехта с говорящим названием “Ретаргетинг коронавируса путем замены эктодомена в шиповидном белке: пересечение видового барьера”:
Исходный текст
Using targeted RNA recombination, we constructed a mutant of the coronavirus mouse hepatitis virus (MHV) in which the ectodomain of the spike glycoprotein (S) was replaced with the highly divergent ectodomain of the S protein of feline infectious peritonitis virus. The resulting chimeric virus, designated fMHV, acquired the ability to infect feline cells and simultaneously lost the ability to infect murine cells in tissue culture.
Кстати, похоже, что Ши Чжэнли стажировалось под руководством Питера Роттиера в Утрехте. По крайней мере, в 2005-ом она была в соавторах совместной статьи, где Утрехт был указан в качестве её аффилиации (но текущий адрес был уже указан в Шанхайском институте). Кстати, сама статья тоже весьма любопытна — в ней авторы исследовали что именно позволяет вирусам расширять свой видовой тропизм:
Исходный текст
Only a relatively few mutations in its spike protein allow the murine coronavirus to switch from a murine-restricted tropism to an extended host range by being passaged in vitro. One such virus that we studied had acquired two putative heparan sulfate-binding sites while preserving another site in the furin-cleavage motif.
Во-первых, любопытно, что фуриновый сайт в том вирусе (SRRAHR|SV) похож на сайт в CoV2 (SPRRAR|SV), хотя у CoV2 он режется более эффективно из-за сдвоенных аргининов (это и делает его polybasic сайтом, то есть у него в последовательности RxxR несколько основных аминокислот подряд):
Но особенно статья любопытна тем, что мутации, позволившие вирусу “расширить свой кругозор”, произошли даже не в лабораторных животных, а в пробирке (by being passaged in vitro). Причем, похоже, происходили они довольно быстро:
Исходный текст
MHV/pi23, a virus obtained after 23 of the 600 passages that resulted in MHV/BHK, also contains a putative HS-binding site in the S1 domain at the same position as in MHV/BHK, albeit as a smaller insertion, while it lacks the putative HS-binding site immediately upstream of the fusion peptide. MHV/pi23 does infect nonmurine cells to some extent but much less efficiently than MHV/BHK. In addition to the multiple HS-binding sites, however, mutations found in other parts of the S protein, such as the HR1 domain and the putative fusion peptide (Fig. 1), might also contribute to the efficient entry into nonmurine cells. We are currently in the process of determining the S protein mutations that are required for the extended host range phenotype.
Забегая вперёд, упомяну, что были и другие группы, которые пытались мутациями в пробирке увеличить вирулентность коронавируса, например MERS-а:
Исходный текст
To better understand the species adaptability of MERS-CoV, we identified a suboptimal species-derived variant of DPP4 to study viral adaption. Passaging virus on cells expressing this DPP4 variant led to accumulation of mutations in the viral spike which increased replication.
Причём у них мутации возникали уже через несколько пассажей (раундов размножения клеточных культур):
(F) Схема появления одиночных и двойных мутаций в шиповидном белке MERS-CoV в разных пассажах.
(G) Расположение мутаций в шиповидном белке MERS-CoV.
Исходный текст
(F) Schematic of single and double mutation emergence in MERS-CoV spike over different passages.
(G) Location of mutations within MERS-CoV spike.
Но это всё будет гораздо позже. А пока давайте вернёмся в 2002 год — ещё ДО вспышки первого SARS-CoV.
Как Барик всем путь озарил
Ральф Барик — это человек-легенда в коронавирусологии. Настоящий первопроходец синтетических методик манипуляции вирусных геномов. Ещё в 2002 году он опубликовал прорывную работу, открывшую новую веху как в изучении различных механизмов природных вирусов, так и в gain-of-function (GOF) исследованиях. В своей работе группа Барика синтетически воссоздала клон природного мышиного коронавируса:
Был разработан новый метод сборки полноразмерной инфекционной кДНК штамма вируса гепатита мыши коронавируса II группы А59 (MHV-A59). Были выделены семь сегментов кДНК, которые охватывали весь геном MHV размером 31,5 тысяч пар нуклеотидов. Концы кДНК были сконструированы с уникальными стыками и собраны только с соседними субклонами кДНК, в результате чего была получена интактная конструкция кДНК MHV-A59 длиной ~ 31,5 т.п.н. Сайты рестриктаз в местах стыков, которые располагались на концах каждого сегмента кДНК, систематически удалялись во время сборки полного полноразмерного продукта кДНК, что позволяет проводить повторную сборку без внесения нуклеотидных изменений… Этот метод потенциально может быть использован для конструирования вирусных, микробных или эукариотических геномов, достигающих нескольких миллионов пар оснований в длину и использоваться для вставки сайтов рестрикции в любом заданном месте в микробном геноме.
Исходный текст
A novel method was developed to assemble a full-length infectious cDNA of the group II coronavirus mouse hepatitis virus strain A59 (MHV-A59). Seven contiguous cDNA clones that spanned the 31.5-kb MHV genome were isolated. The ends of the cDNAs were engineered with unique junctions and assembled with only the adjacent cDNA subclones, resulting in an intact MHV-A59 cDNA construct of?31.5 kb in length. The interconnecting restriction site junctions that are located at the ends of each cDNA are systematically removed during the assembly of the complete full-length cDNA product, allowing reassembly without the introduction of nucleotide changes… The method has the potential to be used to construct viral, microbial, or eukaryotic genomes approaching several million base pairs in length and used to insert restriction sites at any given nucleotide in a microbial genome.
То есть авторы, по сути, перевели РНК-вирус на язык ДНК (с помощью обратной транскриптазы), для того чтобы затем им было удобно манипулировать его геномом с помощью имеющихся инструментов генной инженерии. Создав 7 таких cDNA сегментов провируса, авторы затем их сшили, причем «бесшовно» (не оставляя следов), после чего перевели свою конструкцию обратно в РНК, из которой затем в других клетках сформировались вирусные частицы.
SARS-2003
Буквально спустя несколько недель после публикации той работы группы Барика грянула эпидемия SARS-CoV, и Ральф Барик не терял времени даром. Уже летом 2003 его группа отправила в печать работу по созданию синтетического клона SARS-CoV:
Используя набор смежных сегментов кДНК, которые охватывают весь геном, мы собрали полноразмерную кДНК штамма SARS-CoV Urbani и выделили синтетические клоны вирусы SARS (инфекционный клон SARS-CoV), которые содержали ожидаемые маркерные мутации, вставленные в клоны. Рекомбинантные вирусы реплицировались так же эффективно, как вирус дикого типа, и оба были ингибированы обработкой ингибитором цистеинпротеиназы… Наличие полноразмерной кДНК SARS-CoV создаёт инструмент для манипулирования вирусным геномом, позволяя быстро и рационально разрабатывать и тестировать кандидаты вакцин и терапевтических средств против этого важного человеческого патогена.
Исходный текст
Using a panel of contiguous cDNAs that span the entire genome, we have assembled a full-length cDNA of the SARS-CoV Urbani strain, and have rescued molecularly cloned SARS viruses (infectious clone SARS-CoV) that contained the expected marker mutations inserted into the component clones. Recombinant viruses replicated as efficiently as WT virus and both were inhibited by treatment with the cysteine proteinase inhibitor… Availability of a SARS-CoV full-length cDNA provides a template for manipulation of the viral genome, allowing for the rapid and rational development and testing of candidate vaccines and therapeutics against this important human pathogen.
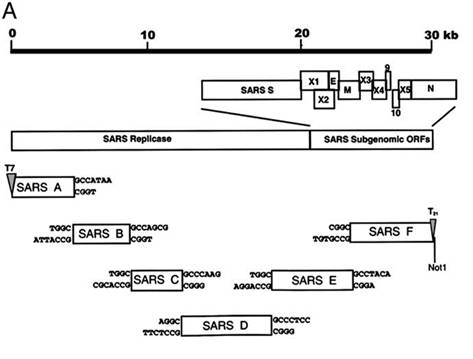
Скорость группы Барика позволяет понять, как быстро квалифицированная команда вирусологов может создать синтетический клон из природного вируса, а значит и вносить в него генетические модификации. Причём это было в 2003 году. Сегодня всё то же самое квалифицированная лаборатория может повторить за считанные недели.
SARS-2006
Барик был первым, но далеко не последним. Генная инженерия развивалась семимильными шагами, создавая всё новые и новые инструменты. Другие группы отрабатывали альтернативные технологии синтетической вирусологии. Например, в 2006 испанские исследователи повторили достижения Барика, тоже создав синтетический клон SARS, но с помощью другой технологии (искусственной бактериальной хромосомы):
Исходный текст
The engineering of a full-length infectious cDNA clone and a functional replicon of the severe acute respiratory syndrome coronavirus (SARS-CoV) Urbani strain as bacterial artificial chromosomes (BACs) is described in this study. In this system, the viral RNA was expressed in the cell nucleus under the control of the cytomegalovirus promoter and further amplified in the cytoplasm by the viral replicase. Both the infectious clone and the replicon were fully stable in Escherichia coli.
…
The assembled SARS-CoV infectious cDNA clone was fully stable during its propagation in E. coli DH10B cells for more than 200 generations, considerably facilitating the genetic manipulation of the viral genome (data not shown). The detailed cloning strategy, plasmid maps, and sequences are available upon request.
Правда, сделали они это не так элегантно как Барик, так как в финальной сборке синтетического вируса у них остались добавленные сайты рестриктаз, в то время как Барик научился соединять фрагменты «бесшовно». Но это мелочи, подход испанцев тоже вполне рабочий — в 2013 году с его помощью они создали синтетический клон MERS-a, а в 2015 году их методика вошла в учебник-справочник по коронавирусам (глава 13):

Ухань-2007
Но вернёмся в 2007 год. Тогда в гонку синтетической вирусологии включилась и группа Ши Чжэнли с работой, изучавшей шиповидный белок человеческого и летучемышиного коронавирусов, пытаясь определить, что именно в нём отвечает за способность перескакивать с вида на вид:
Исходный текст
A series of S chimeras was constructed by inserting different sequences of the SARS-CoV S into the SL-CoV S backbone.
То есть авторы вставляли разные отрезки из белка человеческого SARS-CoV в белок летучемышиного вируса. Вот их вывод:
Исходный текст
From these results, it was deduced that the region from aa 310 to 518 of BJ01-S was necessary and sufficient to convert Rp3-S into a huACE2-binding molecule.
При этом они пробовали заменять и более короткие фрагменты, включая только RBM:
Исходный текст
For introduction of the RBM of SARS-CoV S into the SL-CoV S, the coding region from aa 424 to 494 of BJ01-S was used to replace the corresponding regions of Rp3-S, resulting in a chimeric S (CS) gene designated CS424–494.
С учётом того, что это было написано в 2007 году, думаю, сегодня произвести замену RBM одного вируса на другой не составит труда даже начинающему вирусологу.
Химера-2015
В свете вышеописанных экспериментов, не очень понятно, чем именно был вызван тот фурор, которая произвела, наверное, самая нашумевшая gain-of-function публикация. Речь, конечно же, о совместной работе Ши Чжэнли и Ральфа Барика, в которой они создали синтетический химерный вирус:
Используя систему обратной генетики SARS-CoV, мы создали и охарактеризовали химерный вирус, экспрессирующий шиповидный белок коронавируса летучей мыши SHC014 в мышиноадаптированной основе SARS-CoV. Результаты показывают, что вирусы группы 2b, кодирующие шиповидный белок SHC014 в основе дикого типа, могут эффективно использовать несколько ортологов человеческого рецептора SARS (ACE2), эффективно реплицироваться в первичных клетках дыхательных путей человека и достигать титров in vitro, эквивалентных эпидемическим штаммам SARS-CoV. Кроме того, эксперименты in vivo демонстрируют репликацию химерного вируса в легких мыши с заметным патогенезом. Оценка доступных иммунотерапевтических и профилактических методов на основе атипичной пневмонии показала низкую эффективность; подходы как к моноклональным антителам, так и к вакцинам не смогли нейтрализовать и защитить от инфицирования CoV с новым шиповидным белком. На основании этих результатов мы синтетически воспроизвели инфекционный полноразмерный рекомбинантный вирус SHC014 и продемонстрировали репликацию вируса как in vitro, так и in vivo.
Исходный текст
Using the SARS-CoV reverse genetics system, we generated and characterized a chimeric virus expressing the spike of bat coronavirus SHC014 in a mouse-adapted SARS-CoV backbone. The results indicate that group 2b viruses encoding the SHC014 spike in a wild-type backbone can efficiently use multiple orthologs of the SARS receptor human angiotensin converting enzyme II (ACE2), replicate efficiently in primary human airway cells and achieve in vitro titers equivalent to epidemic strains of SARS-CoV. Additionally, in vivo experiments demonstrate replication of the chimeric virus in mouse lung with notable pathogenesis. Evaluation of available SARS-based immune-therapeutic and prophylactic modalities revealed poor efficacy; both monoclonal antibody and vaccine approaches failed to neutralize and protect from infection with CoVs using the novel spike protein. On the basis of these findings, we synthetically re-derived an infectious full-length SHC014 recombinant virus and demonstrate robust viral replication both in vitro and in vivo.
То есть, по сути, исследователи шли уже проторённой дорогой: взяли шиповидный белок из RsSHC014, который Ши Чжэнли выделила из юньнаньских летучих мышей в 2011 году, и вставили его в SARS-CoV, специально адаптированный под рождённых ползать мышей, для последующих in vivo экспериментов на этих самых мышах. Ну и на человеческих клетках новую конструкцию проверили. А заодно поупражнялись в создании рекомбинантного клона того же самого RsSHC014 — ну а почему бы и нет? Ведь, во-первых, это красиво:
(a) Схема молекулярного клона SHC014-CoV, который был синтезирован в виде шести смежных сегментов кДНК (обозначенных как SHC014A, SHC014B, SHC014C, SHC014D, SHC014E и SHC014F), фланкированных уникальными рестриктазными сайтами BglI, которые обеспечили направленную сборку полноразмерной экспрессии кДНК (для 1a, 1b, спайка, 3, конверт, матрица, 6–8 и нуклеокапсид). Подчеркнутые нуклеотиды представляют собой выступающие последовательности, образованные после расщепления рестриктазой.
Исходный текст
(a) Schematic of the SHC014-CoV molecular clone, which was synthesized as six contiguous cDNAs (designated SHC014A, SHC014B, SHC014C, SHC014D, SHC014E and SHC014F) flanked by unique BglI sites that allowed for directed assembly of the full-length cDNA expressing open reading frames (for 1a, 1b, spike, 3, envelope, matrix, 6–8 and nucleocapsid). Underlined nucleotides represent the overhang sequences formed after restriction enzyme cleavage.
А во-вторых, исследователи поняли, что не только тропностью шиповидного белка к рецептору определяется потенциал вируса к переходу из одного вида животных на другой — потому что химера SHC014-MA15 была более вирулентной чем сам SHC014, даже в человеческих клетках:
Примечательно, что дифференциальный тропизм в легких по сравнению с таковым у SARS-MA15 и ослабление полноразмерного SHC014-CoV в культурах [эпителиальных дыхательных путей человека] относительно SARS-CoV Urbani позволяют предположить, что помимо связывания с ACE2 и другие факторы — включая процессивность шиповидного белка, биодоступность рецептора или антагонизм иммунных ответов хозяина — могут способствовать вирулентности.
Исходный текст
Notably, differential tropism in the lung as compared to that with SARS-MA15 and attenuation of full-length SHC014-CoV in [human epithelial airway cell] cultures relative to SARS-CoV Urbani suggest that factors beyond ACE2 binding — including spike processivity, receptor bio-availability or antagonism of the host immune responses — may contribute to emergence.
Особо хочу выделить процессивность шиповидного белка в цитате, потому что это далеко не первый раз когда исследователи писали, что способность шиповидного белка к расщеплению протеазами (включая фурин) оказывает значительное влияние на вирулентность.
В заключение темы — совместное фото Ральфа Барика и Ши Чжэнли. Фото сделано в Ухане, в октябре 2018-го:

Мышиный SARS-2007
Тут не могу не упомянуть, что это был за “мышиный вирус MA15” в предыдущей работе. Это вовсе не какой-то природный мышиный коронавирус, как можно было бы предположить. А это лабораторно модифицированный человеческий SARS-CoV, который ещё в 2007-о<