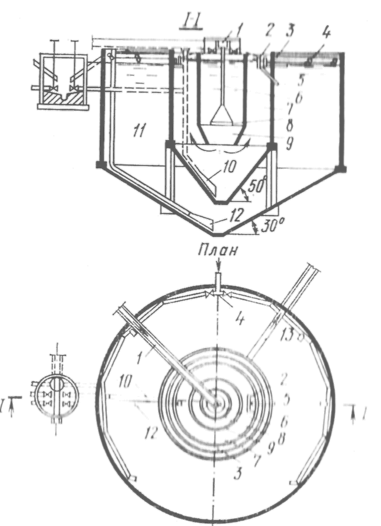
Установка замедленного коксования: Чем выше температура и ниже давление, тем место разрыва углеродной цепи всё больше смещается к её концу и значительно возрастает...
Интересное:
Берегоукрепление оползневых склонов: На прибрежных склонах основной причиной развития оползневых процессов является подмыв водами рек естественных склонов...
Искусственное повышение поверхности территории: Варианты искусственного повышения поверхности территории необходимо выбирать на основе анализа следующих характеристик защищаемой территории...
I. Моногенные болезни: аутосомно-доминантные, аутосомно-рецессивные и сцепленные с полом.
Задача медицинской генетики: выявление молекулярной природы мутации и первичного молекулярного дефекта.
Название болезни
Фото
Тип наследования, частота встречаемости
Первичный молекулярный дефект
Первичный биохимический дефект
Энзимопатии. Болезни аминокислотного обмена.
Фенилкетонурия
Аутосомно-рецессивный
1:10.000
Мутация гена 12q22-24, который контролирует синтез фермента фенилаланингидроксилазы. Известно более 30 мутаций в этом гене.
Недостаточность фермента фенилаланингидроксилазы. Этот фермент отвечает за превращение фенилаланин в тирозин. Фенилаланин - аминокислота, которая поступает в организм с пищей.
Фенилкетонурия – болезнь накопления.
Характеристика. Недостаточность фермента фенилаланингидроксилазы приводит к накоплению в крови аминокислоты фенилаланина. Ребенок рождается фенотипически здоровым. С началом грудного вскармливания постепенно развивается олигофрения. В моче обнаруживаются фенилпировиноградная и фенилуксусная кислоты, а в крови отмечается повышение содержания фенилаланина. Характерен «мышиный» запах мочи. Слабая пигментация кожи, волос, радужной оболочки глаз. Если больного ребенка кормить пищей без фенилаланина, можно предотвратить развитие олигофрении. Для диагностики используют реакцию Фелинга (реактив FeCl3 – сине-зеленая окраска) или реакцию с 2,4-динитрофенилгидразином (ярко-желтый оттенок).
Тирозиноз
Аутосомно-рецессивный
Мутации в гене, который кодирует оксидазу гидроксифенилпировиноградной кислоты.
Накапливается тирозин и гидроксифенилпировиноградная кислота. Болезнь накопления.
Характеристика. Задержка психо-моторного развития. Цирроз печени. Изменяется костная система. Тяжелые формы без лечения приводят к смерти в 6-7 месяцев.
Альбинизм
Аутосомно-рецессивный.
1:20.000-1:39.000
Мутации в гене, кодирующем фермент тирозиназу.
Отсутствие фермента тирозиназы приводит к нехватке меланина.
Болезнь недостатка.
Характеристика. Отсутствие пигмента в коже, в волосах и радужной оболочке глаз (у любой расы). Кожа больных красная, не загорает на солнце. Радужка глаз серо-голубого цвета, волосы белые или желтые.
Алькаптонурия
Аутосомно-рецессивный. 2-5 больных на 1 млн. новорожденных
Мутации в гене, который кодирует фермент оксидазу гомогентизиновой кислоты.
Фермент гипоксантингуанин фосфорибозил трансфераза катализирует превращение свободных пуриновых оснований (гуанина и гипоксантина) в нуклеотиды. При недостаточной активности этого фермента наблюдается повышенная экскреция с мочой мочевой кислоты и уратов. Спастические церебральные параличи. Умственная отсталость. Отставание в моторном развитии. Приступы агрессивного поведения со склонностью к членовредительству (аутоагрессия). Гематурия. Камни в мочевыводящих путях. Нефропатия. Острый подагрический артрит (поражение суставов вследствие отложения солей мочевой кислоты). Проявляется только у мальчиков.
Гемоглобинопатии.
Серповидно-клеточная анемия
Аутосомно-доминантный (с неполным доминированием)
Миссенс-мутация (замена нуклеотида, приводящая к замене аминокислоты) в гене, кодирующем β-цепь гемоглобина.
В 6-м положении β-цепи гемоглобина (Hb) глутаминовая кислота заменяется на валин. У гетерозигот измененный Hb составляет 20-45%, у гомозигот 60-99% общего Hb.
Нормальный Hb А превращается в Hb S, который в условиях недостатка кислорода полимеризуется, образуя кристаллы и волокна. Гомозиготы погибают, не достигнув зрелого возраста. В условиях низкого парциального давления кислорода мутантные эритроциты приобретают форму серпа. Такие эритроциты теряют гибкость. Происходит образование тромбов, особенно в костях. Возникают костные боли. Наблюдаются анемия (бледная кожа), увеличение печени и селезенки (гепатоспленомегалия). Снижение работоспособности. Гетерозиготы устойчивы к малярии.
Талассемия
Аутосомно-доминантный (с неполным доминированием)
Мутации в генах, кодирующих α-цепь и β-цепь гемоглобина
α-талассемия характеризуется той или иной степенью дефицита α-глобиновых цепочек при нормальной продукции β-глобинов. Избыток β-цепочек приводит к формированию тетрамеров β4. Такой гемоглобин (HbH) значительно снижает способность переносить кислород. Гетерозиготы устойчивы к малярии.
Синдром Марфана
Аутосомно-доминантный.
Мутации в гене фибриллина.
Нарушение в синтезе и структуре фибриллина.
Белок соединительной ткани фибриллин участвует в формировании волокон коллагена из проколлагена. Тонкие и длинные «паучьи пальцы», нарушения в строении хрусталика, может наблюдаться катаракта. Происходят изменения со стороны сердечно-сосудистой и других систем организма. Деформация позвоночника, вдавленная грудная клетка.
Хорея Гентигтона
Аутосомно-доминантный
Динамическая мутация – экспансия триплетов. Увеличение числа повторов (СAG)n между промотором и структурной частью гена. Если n=9-32 – норма, 33-44 - предмутация, 45-100 – развивается заболевание. Частота 1:10000 – 100000.
Дегенеративные изменения головного мозга, нарастающие движения лица и конечностей, затрудненная речь, прогрессирующее слабоумие (деменция). Признаки заболевания проявляются, как правило в возрасте 35-40 лет и позже. Манифестация заболевания увеличивается с возрастом. Передача мутантного аллеля от отца сопровождается более тяжелой клиникой. У гомозигот болезнь проявляется тяжело.
Муковисцидоз
Аутосомно-рецессивный.
1:2000
Мутация гена, который кодирует хлорный канал. Наблюдается делеция трех нуклеотидов, в результате которой в транспортном белке отсутствует 1 аминокислота.
Проявлением болезни является нарушение транспорта ионов Cl- и Na+ в эпителиальных клетках поджелудочной железы, бронхов, кишечника и других органов.
Избыточное выведение хлоридов приводит к большой секреции слизи. Выводные протоки желез закупориваются, вязкая слизь не выводится. Возникают кисты поджелудочной железы, закупорка мелких бронхов. При диагностике учитывается повышение концентрации Cl- и Na+.
Добавить эритроцитоз
Генные болезни
I. Моногенные болезни: аутосомно-доминантные, аутосомно-рецессивные и сцепленные с полом.
Задача медицинской генетики: выявление молекулярной природы мутации и первичного молекулярного дефекта.
Название болезни
Фото
Тип наследования, частота встречаемости
Первичный молекулярный дефект
Первичный биохимический дефект
Энзимопатии. Болезни аминокислотного обмена.
Фенилкетонурия
Аутосомно-рецессивный
1:10.000
Мутация гена 12q22-24, который контролирует синтез фермента фенилаланингидроксилазы. Известно более 30 мутаций в этом гене.
Недостаточность фермента фенилаланингидроксилазы. Этот фермент отвечает за превращение фенилаланин в тирозин. Фенилаланин - аминокислота, которая поступает в организм с пищей.
Фенилкетонурия – болезнь накопления.
Характеристика. Недостаточность фермента фенилаланингидроксилазы приводит к накоплению в крови аминокислоты фенилаланина. Ребенок рождается фенотипически здоровым. С началом грудного вскармливания постепенно развивается олигофрения. В моче обнаруживаются фенилпировиноградная и фенилуксусная кислоты, а в крови отмечается повышение содержания фенилаланина. Характерен «мышиный» запах мочи. Слабая пигментация кожи, волос, радужной оболочки глаз. Если больного ребенка кормить пищей без фенилаланина, можно предотвратить развитие олигофрении. Для диагностики используют реакцию Фелинга (реактив FeCl3 – сине-зеленая окраска) или реакцию с 2,4-динитрофенилгидразином (ярко-желтый оттенок).
Тирозиноз
Аутосомно-рецессивный
Мутации в гене, который кодирует оксидазу гидроксифенилпировиноградной кислоты.
Накапливается тирозин и гидроксифенилпировиноградная кислота. Болезнь накопления.
Характеристика. Задержка психо-моторного развития. Цирроз печени. Изменяется костная система. Тяжелые формы без лечения приводят к смерти в 6-7 месяцев.
Альбинизм
Аутосомно-рецессивный.
1:20.000-1:39.000
Мутации в гене, кодирующем фермент тирозиназу.
Отсутствие фермента тирозиназы приводит к нехватке меланина.
Болезнь недостатка.
Характеристика. Отсутствие пигмента в коже, в волосах и радужной оболочке глаз (у любой расы). Кожа больных красная, не загорает на солнце. Радужка глаз серо-голубого цвета, волосы белые или желтые.
Алькаптонурия
Аутосомно-рецессивный. 2-5 больных на 1 млн. новорожденных
Мутации в гене, который кодирует фермент оксидазу гомогентизиновой кислоты.
Поперечные профили набережных и береговой полосы: На городских территориях берегоукрепление проектируют с учетом технических и экономических требований, но особое значение придают эстетическим...
Археология об основании Рима: Новые раскопки проясняют и такой острый дискуссионный вопрос, как дата самого возникновения Рима...