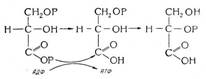
Г-6-ФДГ - фермент обмена углеводов, большое количество фермента содержится в эритроцитах. Этот фермент необходим для поддержания нормального внутриклеточного содержания глютатиона, защищающего гемоглобин и мембрану эритроцита от окисления. Недостаточность фермента приводит к разрушению мембран и выпадению в осадок цепей глобина, а также повышенному содержанию эритроцитов. При отсутствии Г-6-ФДГ в эритроцитах происходит нарушение функционирования гемоглобина.
Глюкозо-6-фосфатдегидрогеназа в эритроцитах - показатель ферментопатии (нарушение образования фермента), приводящей к развитию гемолитической анемии.
Врожденный дефицит Г-6-ФДГ эритроцитов относится к распространенным наследственным аномалиям (энзимопатиям). Дефицит Г-6-ФДГ наследуется по рецессивному типу, сцепленному с полом, в связи с чем клинические проявления данной патологии наблюдаются преимущественно у мужчин.
Айсель
61.Микросфероцитарная гемолитическая анемия (болезнь Минковского–Шоффара)
Как известно, для нормального функционирования эритроцита необходимо поддержание его нормальной формы, иначе эритроцит просто не пройдёт через просвет сосуда. В поддержании двояковогнутой формы эритроцита участвует белок спектрин. При анемии Минковского-Шоффара имеется дефект синтеза данного белка. К тому же происходит нарушение работы АТФ-азы, ввиду чего возникает повышенное поступление натрия и воды в клетку. Это приводит к тому, что эритроцит приобретает форму сферы и имеет маленький размер. Из-за этого резко падает способность эритроцита восстанавливать свою структуру после прохождения сосудов, поэтому когда эти клетки крови проходят межсинусные пространства селезёнки, те из них, которые имеют патологическую форму, разрушаются. Из-за этого вместо 120 дней при наследственном микросфероцитозе эритроцит живет всего лишь 10.
62. Серповидноклеточная анемия
Серповидноклеточная анемия- патология гемоглобина. Мутация гена hbb приводит к синтезу аномального гемоглобина S. В условия гипоксии гемоглобин S полимеризуется и откладывается в виде длинных цепей, изменяющих форму эритроцита(серповидность). Эти эритроциты блокируют систему циркуляции и приводят к развитию инфарктов разных органов.
63.Талассемии
Талассемии- гипохромные микроцитарные гемолитические анемии, обусловленные нарушением синтеза альфа- или бета- глобина.
Альфа-талассемия- у новорожденных избыточный синтез гамма- глобина и образование гемоглобина Барта. Нестабильно и в 10 раз сильнее связывают кислород и не могут его транспортировать. Быстрое старение и разрушение эритроцитов. Гомозиготная альфа-талассемия при делении 4 генов несовместима с жизнью. Смерть внутриутробная или в первые часы после рождения.
Бета-талассемия-избыток альфа- и недостаток бета-цепей. разрушение эритрокариоцитов в костном мозге и неэффективный гемопоэз. Усиленное разрушение в селезенке, спленомегалия. Аномалии скелета. Отставание в росте.
Анемия Дайемонда-Блекфена (вариант анемии Фанкоми)-врожденная форма красноклеточной аплозии. Низкое содержание ретикулоцитов и эритрокариоцитов в костном мозге.
64.Агранулоцитоз наследственный (син. - врождённая нейтропения, наследуемый агранулоцитоз новорождённых, болезнь Костманна)
Различные генные дефекты,значительно ухудшающие действие нейтрофилов, зарегистрированы при синдромах Йома, Костманна, Шедьяки- Хигаши, синдроме ленивых лейкоцитов, недостаточности миелопероксидазы и катепсина G.
Агранулоцитоз наследственный- характерные признаки наследуемых агранулацитов(нейтропении)- лихорадка, рекуррентные инфекции кожных покровов и дыхательных путей. Симптоматика определяется формой заболевания.
Болезнь Костмана- (в т.ч. дефет рецептора колониестимулирущего фактора гранулоцитов [ген С3F3R],р, ассоциация с экспрессией HLA-B12). Тяжелые и часто смертельные рекуррентные инфекции кожных покровов (фурункулез, рожистое воспаление) и дыхательных путей наблюдают с первых месяцев жизни. В костном мозг обнаруживают задержку созревания нейтрофилов на стадии промиелоцита или юного миелоцита. Трансплантация костного мозга.
Синонимы: врожденная нейтропения, наследуемый агранулоцитоз новорожденных.
65.Дефекты функций нейтрофилов (синдромы Йова, Костманна, Шедьяка-Хигаши, ленивых лейкоцитов и др.)
- Иммунодефицит-Дефициты фагоцитоза- синдром Йова
- Первичный дефект фагоцитоза, частичный иммунодефицит, Патологические изменения гранул и ядер всех типов лейкоцитов, альбинизм, возможна гиперпигментация кожи, анемия, тромбоцитопения, изменения в костях, лёгких, сердце, а также психомоторные дефекты и предрасположенность к инфекциям.- синдром Шедьяка-Хигаши
- Ограниченная подвижность гранулоцитов и нарушение их выхода из костного мозга в кровь- синдром ленивых лейкоцитов
Диана
66. Тромбоцитопатии (тромбастения Глянцманна, синдром Бернара—Сулье, серых тромбоцитов синдром)
Тромбастения Гланцмана – наследственная болезнь из группы геморрагических диатезов, характеризующаяся недостаточностью ряда ферментов в тромбоцитах, вторичным нарушением ретракции кровяного сгустка и часто удлиненным временем кровотечения при нормальном или слегка пониженном количестве тромбоцитов, наличием гигантских тромбоцитов; выделяют формы с аутосомно-рецессивным и аутосомно-доминантным типом наследования.
Для тромбастении Гланцмана характерно возникновение гематом, кровотечений из слизистых и гиперменореи, а также тяжелых кровотечений в ответ на нормальные стимулы (например, хирургическое вмешательство или травму).
Синдром Бернара-Сулье - врожденная тромбоцитопатия, сопровождающаяся кровоизлияниями в кожу, слизистые оболочки, внутренние органы, развитием носовых кровотечений
Синдром серых тромбоцитов (недостаточность а-гранул): увеличение размеров тромбоцитов, серая окраска. снижение количества а-гранул и тромбоцитспецифических белков а-гранул.
67. Наследственные коагулопатии
Коагулопатии
собирательное обозначение болезненных состояний, обусловленных нарушениями физиологических механизмов свертывания крови, приводящие обычно к развитию геморрагического синдрома.
Наследственные коагулопатии вызываются генетически обусловленным снижением или извращением плазменных компонентов гемостаза. Наиболее распространенными формами являются гемофилия А, В, С, афибриногенемия.
Наследственные коагулопатии делятся на:
- Гемофилии: А-дефицит VIII фактора, В-дефицит IX фактора, С-дефицит XI фактора, Д-дефицит XII;
- Парагемофилия: дефицит II, V, VII, X факторов;
- Нарушение образования фибрина, дефицит фибриногена (I фактора).
68.Болезнь фон Виллебранда
наследственное заболевание крови, характеризующееся возникновением эпизодических спонтанных кровотечений, которые схожи с кровотечениями при гемофилии. Заболевание наследуется по принципу аутосомного доминирования.
Причина кровотечений — нарушение свертываемости крови из-за недостаточной активности фактора Виллебранда.
Наиболее характерным и специфическим симптомом при болезни Виллебранда являются кровотечения из
слизистых полости рта, носа, внутренних органов.
Надя