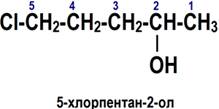Спирты
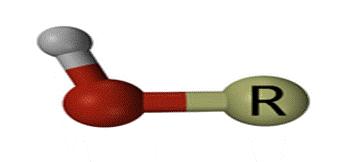
Отличительная особенность спиртов — гидроксильная группа при насыщенном атоме углерода — на рисунке выделена красным (кислород) и серым цветом (водород).
Спирты́ (устар. алкого́ли, англ. alcohols; от лат. spiritus — дух) — органические соединения, содержащие одну или более гидроксильных групп (гидроксил, − O H), непосредственно связанных с насыщенным (находящемся в состоянии sp³ гибридизации) атомом углерода. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водорода замещен на органическую функциональную группу: R−OH.
В номенклатуре IUPAC для соединений, в которых гидроксильная группа связана с ненасыщенным (находящемся в состоянии sp2—гибридизации атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C связью) и «фенолы» (гидроксил связан с бензольным или другим ароматическим циклом).
Спирты являются обширным и очень разнообразным классом органических соединений: они широко распространены в природе, имеют важнейшее промышленное значение и обладают исключительными химическими свойствами.
По мнению издания The 100 Most Important Chemical Compounds (Greenwood Press, 2007), в сотне самых важных химических соединений четыре позиции занимают спирты (холестерин, этанол, глицерин и метанол).
Классификация спиртов
Спирты классифицируются следующим образом:
· по числу гидроксильных групп
| — одноатомные спирты (метанол);
— двухатомные спирты (этиленгликоль);
— трехатомные спирты (глицерин);
— четырёхатомные спирты (пентаэритрит);
— многоатомные спирты (пятиатомный спирт: ксилит).
| 
| 
|
| трёхатомный спирт глицерин
| четырехатомный спирт пентаэритрит
|
· в зависимости от насыщенности углеводородного заместителя
| — предельные или насыщенные спирты (бутанол); — непредельные или ненасыщенные спирты (аллиловый спирт, пропаргиловый спирт); — ароматические спирты (бензиловый спирт);
| |
· в зависимости от наличия или отсутствия цикла в углеводородном заместителе
| — алициклические спирты (циклогексанол); — алифатические или ациклические спирты (этанол);
| |
· в зависимости от того, при каком атоме углерода находится гидроксильная группа
| — первичные спирты (пропанол); — вторичные спирты (изопропиловый спирт); — третичные спирты (2-метилпропан-2-ол).
| 
|
Номенклатура спиртов
Систематическая номенклатура
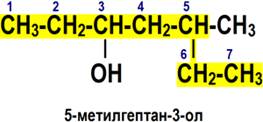
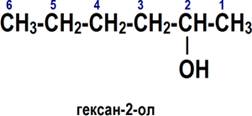
По номенклатуре ИЮПАК названия простых спиртов образуются от названий соответствующих алканов с добавлением суффикса «-ол», положение которого указывается арабской цифрой.
Правила построения названия спиртов (функциональная группа −OH):
1. Выбирается родительский углеводород по самой длинной непрерывной углеводородной цепи, содержащей функциональную группу. Он формирует базовое название (по числу атомов углерода)
2. Родительский углеводород нумеруется в направлении, которое дает суффиксу функциональной группы самое низкое число.
3. Если в соединении помимо функциональной группы имеется другой заместитель, суффикс функциональной группы получает самое низкое число.
4. Если для суффикса функциональной группы получено одно и то же число в обоих направлениях, цепь нумеруется в направлении, которое дает другому заместителю самое низкое число.
5. Если имеется несколько заместителей, они перечисляются в алфавитном порядке.
Имена заместителей ставятся перед именем родительского углеводорода, а суффикс функциональной группы — после. ИЮПАК рекомендует цифру, характеризующую положение функциональной группы, писать сразу после имени углеводородного заместителя перед суффиксом функциональной группы.
Для многоатомных спиртов перед суффиксом -ол по-гречески (-ди-, -три-, …) указывается количество гидроксильных групп (например: пропан-1,2,3-триол).
Рациональная и тривиальная номенклатура
Спирты, даже в научной литературе, часто называются по правилам, отличным от современной номенклатуры ИЮПАК. Очень распространенным является образование названия, как производного от соответствующего алкана, преобразованного в прилагательное с добавлением слова спирт
· этан С2Н6 → эт иловый спирт C2H5OH;
· гексан С6Н14 → гекс иловый спирт C6H13OH.
Рациональная номенклатура спиртов рассматривает их как производные метанола CH3OH или по другому карбинола
· (СH3)2CНOH → диметилкарбинол;
· (С6H5)3COH → трифенилкарбинол.
В популярной и научной литературе можно нередко встретить исторические или тривиальные названия спиртов, которые вследствие сложившейся традиции используются вместо официальной химической терминологии.
Систематические и тривиальные названия некоторых спиртов приведены в табл. 1. 1 Систематические и тривиальные названия некоторых спиртов
| Химическая формула спирта
| Название по номенклатуре ИЮПАК
| Тривиальное название
|
| Предельные одноатомные спирты
|
| CH3OH
| Метанол
| Древесный спирт
|
| C2H5OH
| Этанол
| Винный спирт
|
| C5H11OH
| Пентан-1-ол
| Амиловый спирт
|
| C16H33OH
| Гексадекан-1-ол
| Цетиловый спирт
|
| Предельные двух-, трех-, четырёхатомные спирты
|
| C2H4(OH)2
| Этан-1,2-диол
| Этиленгликоль
|
| C3H5(OH)3
| Пропан-1,2,3-триол
| Глицерин
|
| C5H8(OH)4
| 2,2- бис (Гидроксиметил)пропан-1,3-диол
| Пентаэритрит
|
| Предельные многоатомные спирты
|
| C5H7(OH)5
| Пентан-1,2,3,4,5-пентол
| Ксилит
|
| C6H8(OH)6
| Гексан-1,2,3,4,5,6-гексол
| Маннит, Сорбит
|
| Непредельные алифатические спирты
|
| C3H5OH
| Проп-2-ен-1-ол
| Аллиловый спирт
|
| C10H17OH
| 3,7-диметилокта-2,7-диен-1-ол
| Гераниол
|
| C3H3OH
| Проп-2-инол-1
| Пропаргиловый спирт
|
| Алициклические спирты
|
| C6H6(OH)6
| Циклогексан-1,2,3,4,5,6-гексол
| Инозит
|
| C10H19OH
| 2-(2-пропил)-5-метилциклогексанол-1
| Ментол
|
Этимология
Слово алкого́ль происходит из арабского языка الكحل (al-ku ḥ l) и означает «порошкообразная сурьма». В русский язык слово пришло через нем. Alkohol, нидерл. alkohol или порт., исп. alcohol. Однако в русском языке сохранился в виде архаизма, по всей видимости, и омоним слова «алкоголь» в значении «мелкий порошок».[Слово спирт появилось в русском языке во времена Петра I через английское слово spirit, которое, в свою очередь, произошло от латинского spīritus — «дыхание, дух, душа».
История открытия спиртов
Этиловый спирт, вернее, хмельной растительный напиток, его содержащий, был известен человечеству с глубокой древности. Считается, что не менее чем за 8000 лет до нашей эры люди были знакомы с действием перебродивших фруктов, а позже — с помощью брожения получали хмельные напитки, содержащие этанол, из фруктов и мёда. Археологические находки свидетельствуют, что в Западной Азии виноделие существовало ещё в 5400—5000 годах до н.э., а на территории современного Китая, провинция Хэнань, найдены свидетельства производства «вина», вернее ферментированных смесей из риса, мёда, винограда и, возможно, других фруктов, в эпоху раннего неолита: от 6500 до 7000 гг. до н.э.
Впервые спирт из вина получили в VI—VII веках арабские химики, а первую бутылку крепкого алкоголя (прообраза современной водки) изготовил персидский алхимик Ар-Рази в 860 г. В Европе этиловый спирт был получен из продуктов брожения в XI—XII веке, в Италии. В Россию спирт впервые попал в 1386 г., когда генуэзское посольство привезло его с собой под названием «аква вита» и презентовало царскому двору.
В 1660 г. английский химик и богослов Роберт Бойль впервые получил обезвоженный этиловый спирт, а также открыл его некоторые физические и химические свойства, в частности обнаружив способность этанола выступать в качестве высокотемпературного горючего для горелок. Абсолютированный спирт был получен в 1796 г. русским химиком Т. Е. Ловицем. В 1842 г. немецкий химик Я. Г. Шиль открыл, что спирты образуют гомологический ряд, отличаясь на некоторую постоянную величину. Правда, он ошибся, описав её как C2H2. Спустя два года, другой химик Шарль Жерар установил верное гомологическое соотношение CH2 и предсказал формулу и свойства неизвестного в те годы пропилового спирта. В 1850 г. английский химик Александр Вильямсон, исследуя реакцию алкоголятов с иодистым этилом, установил, что этиловый спирт является производным от воды с одним замещенным водородом, экспериментально подтвердив формулу C2H5OH. Впервые синтез этанола действием серной кислоты на этилен осуществил в 1854 г. французский химик Марселен Бертло.
Первое исследование метилового спирта было сделано в 1834 г. французскими химиками Жаном-Батистом Дюма и Эженом Пелиго (англ.); они назвали его «метиловым или древесным спиртом», так как он был обнаружен в продуктах сухой перегонки древесины. Синтез метанола из метилхлорида осуществил французский химик Марселен Бертло в 1857 г.. Им же был открыт в 1855 г. изопропиловый спирт, действием на пропилен серной кислотой.
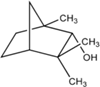
Впервые третичный спирт (2-метил-пропан-2-ол) синтезировал в 1863 г. известный русский ученый А. М. Бутлеров, положив начало целой серии экспериментов в этом направлении. Двухатомный спирт — этиленгликоль — впервые был синтезирован французским химиком А.Вюрцем в 1856 г.. Трехатомный спирт — глицерин — был обнаружен в природных жирах ещё в 1783 г. шведским химиком Карлом Шееле, однако его состав был открыт только в 1836 г., а синтез осуществлен из ацетона в 1873 г. Шарлем Фриделем.
Нахождение в природе
Спирты имеют самое широкое распространение в природе, особенно в виде сложных эфиров, однако и в свободном состоянии их можно встретить достаточно часто. Метиловый спирт в небольшом количестве содержится в некоторых растениях, например: борщевике (Heracleum).
Этиловый спирт — естественный продукт спиртового брожения органических продуктов, содержащих углеводы, часто образующийся в прокисших ягодах и фруктах без всякого участия человека. Кроме того, этанол является естественным метаболитом и содержится в тканях и крови животных и человека. В эфирных маслах зелёных частей многих растений содержится 3(Z)-Гексен-1-ол («спирт листьев»), придающий им характерный запах. Фенилэтиловый спирт — душистый компонент розового эфирного масла. Очень широко представлены в растительном мире терпеновые спирты, многие из которых являются душистыми веществами
| 
| 
| 
| 
|
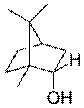
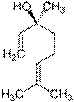
| Борнеол — в древесине борнеокамфорного дерева
| Ментол — содержится в эфирном масле мяты и герани.
| Гераниол — содержится во многих эфирных цветочных маслах
| Линалоол — содержится во многих цветочных эфирных маслах
| Цитронеллол — содержится во многих эфирных маслах
|
| 
| 
| 
| 
| 
|
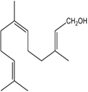
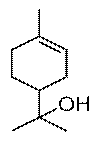
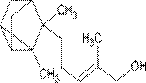
| Фарнезол — содержится во многих эфирных цветочных маслах
| Терпинеол — содержится во многих эфирных маслах
| Бисаболол — входит в состав эфирного масла ромашки, тополя
| Санталол — входит в состав древесины сандалового дерева
| Фенхол — содержится в смоле хвойных деревьев и плодах фенхеля
|

| 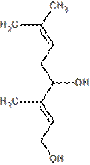
| 
| 
| 
|
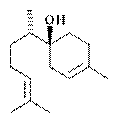
| Нерол — содержится в эфирных маслах многих цветов
| Розиридол — содержится в эфирном масле родиолы розовой
| Туйол — содержится в эфирном масле шалфея
| Сабинол — содержится в эфирном масле тысячелистника
| Хризантемиловый спирт — в эфирном масле полыни однолетней
|
| | | | | | | | | |
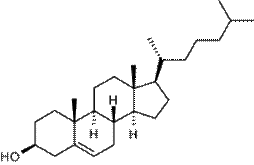
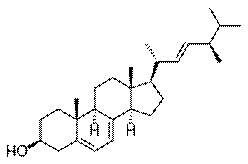
В животном и растительном мире распространены конденсированные тетрациклические спирты (производные гонана), обладающие высокой биологической активностью и входящие в класс стероидов
| 
|
| Холестерол (холестерин) — содержится в клетках практически всех живых организмов, особенно животных
| Эргостерол — содержится в некоторых водорослях и грибах
|
Отдельную группу стероидов составляют жёлчные многоатомные спирты, находящиеся в жёлчи животных и человека: буфол, холестантетрол, холестанпентол, миксинол, сцимнол, химерол и пр. В природе находятся разнообразные многоатомные или сахарные спирты
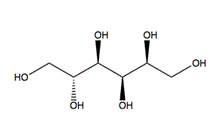
| 
|
| Сорбит — содержится в ягодах вишни и рябины
| Маннит — содержится в морских водорослях, грибах
|
Энергия связей в метаноле
| Энергия связи, кДж/моль
|
| Водородная связь
| Связь С—H
| Связь С—O
| Связь O—H
|
| 16,7
| 391,7
| 383,5
| 428,8
|
Молекулы спирта, имея две полярных связи C−O и O−H, обладают дипольным моментом (для алканолов: 5,3-6,0·10−30 Кл·м). Электростатические заряды в молекуле метанола составляют: на атоме углерода 0,297 e; на атоме гидроксильного водорода 0,431 e; на атоме кислорода −0,728 e. Вместе с тем, энергия ионизации спиртов ниже, чем у воды, что объясняется электронодонорным эффектом алкильной группы
· Вода: 12,61 эВ;
· Метиловый спирт: 10,88 эВ;
· Этиловый спирт: 10,47 эВ;
· Изопропиловый спирт: 10,12 эВ;
· Аллиловый спирт: 9,67 эВ.
Следует отметить, что влияние гидроксильной группы особенно велико на соединения с небольшой углеводородной цепочкой. Так, например, метанол и этанол неограниченно смешиваются с водой и имеют довольно высокие плотности и температуры кипения для своей молекулярной массы, в то время как высшие спирты гидрофобны и мало отличаются по свойствам от соответствующих углеводородов (табл. 4.).
Химические свойства спиртов
Физико-химические свойства спиртов определяются в основном строением углеводородной цепи и функциональной группы −OH, а также их взаимным влиянием:
1) чем больше заместитель, тем сильнее он влияет на функциональную группу, снижая полярность связи O—Н. Реакции, основанные на разрыве этой связи, протекают более медленно;
2) гидроксильная группа −ОН уменьшает электронную плотность вдоль прилегающих σ-связей углеродной цепи (отрицательный индуктивный эффект).
Все химические реакции спиртов можно разделить на три условных группы, связанных с определёнными реакционными центрами и химическими связями:
· разрыв связи O−H (реакционный центр — водород);
· разрыв или присоединение по связи С−OH (реакционный центр — кислород);
· разрыв связи −СOH (реакционный центр — углерод).
Реакции с участием гидроксильной группы (связи С−O и О−H)
Кислотно-основные реакции спиртов
Со щелочными и щелочноземельными металлами, алюминием, галлием, таллием и некоторыми другими металлами, а также сильными основаниями (например: амидами или гидридами) спирты способны реагировать с образованием алкоголятов
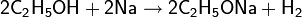

С сильными кислотами Льюиса спирты ведут себя подобно основаниям, образуя донорно-акцепторные комплексы
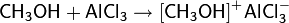
Превращение спиртов в галогеналканы
Одной из наиболее важных реакций с участием связи C−O является превращение спиртов в галогеноалканы. Гидроксильная группа в спиртах может быть замещена на атом галогена несколькими способами:
· взаимодействием с галогенводородами (HCl, HBr, HI);
· реакцией с тионилхлоридом;
· действием галогенидов фосфора (III) и (V);
· реакцией с квазифосфониевыми солями;
· превращением в алкилсульфонат с последующей реакцией замещения.
Взаимодействие спиртов с галогенводородами
Взаимодействие спиртов с галогенводородными кислотами приводит к замещению гидроксильной группы на галоген
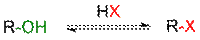
В зависимости от строения субстрата возможны побочные процессы изомеризации и дегидратации. Из-за относительно жестких условий проведения данные реакции применимы только к соединениям, устойчивым к кислотам.
Бромоводородную и иодоводородную кислоты часто получают непосредственно в ходе реакции из соответствующих солей (KBr, KI и т.д.) действием серной или фосфорной кислот. Незамещенные первичные спирты превращаются в алкилбромиды с помощью горячей концентрированной бромоводородной кислоты
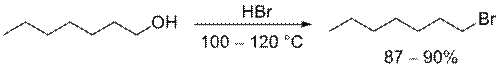
Попытки получить алкилиодид с помощью HI иногда могут приводить к восстановлению первоначального продукта до алкана. Помимо этого, свободный иодоводород способен реагировать с серной кислотой, приводя к образованию сернистой кислоты и иода. Если субстрат содержит двойные связи, последние также могут быть восстановлены
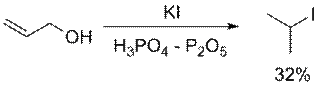
Вышеуказанные реакции можно использовать для получения первичных, вторичных и третичных галогеноалканов, хотя в случае изобутилового и неопентилового спиртов велики выходы продуктов перегруппировки
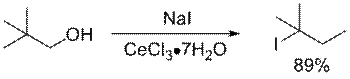
Реакции третичных спиртов с HCl протекают достаточно легко. При этом образуются соответствующие третичные алкилхлориды (совместно с продуктами побочных реакций). Первичные и вторичные спирты реагируют гораздо медленнее и требуют применения катализатора. Обычно используется так называемый реагент Лукаса, представляющий собой смесь HCl и ZnCl2
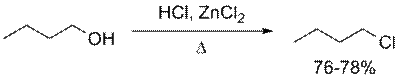
Хорошие выходы первичных алкилхлоридов были также получены при использовании HCl в HMPA (гексаметилфосфотриамид, биполярный апротонный растворитель).
Прямое взаимодействие спиртов с фтороводородом возможно только при использовании третичных, аллиловых и бензиловых спиртов. Так, например, реакция трет -бутилового спирта c 60 % водным раствором HF при нагревании приводит к образованию трет -бутилфторида
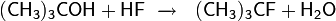
Вместо чистой HF для фторирования обычно используют 70 % раствор фтороводорода в пиридине, так называемый реактив Олаха. Первичные и вторичные спирты реагируют с галогенводородами по механизму SN2 (общая схема):
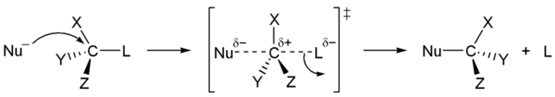
Для третичных спиртов характерен механизм SN1
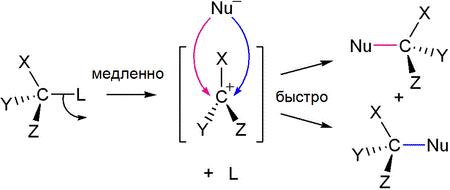
В ходе такого замещения образуется промежуточный карбокатион, поэтому SN1 реакции могут сопровождаться перегруппировками и элиминированием. Таким образом, практический интерес представляют только те третичные спирты, которые дают карбокатион, не способный к перегруппировкам.
Взаимодействие спиртов с галогенидами фосфора
Распространённым способом превращения спиртов в алкилгалогениды является их взаимодействие с галогенидами фосфора: РВr3, РСl5, РОСl3 или РI3 (образуется непосредственно в ходе реакции). Реакция протекает по нуклеофильному механизму с образованием галогенфосфита в качестве интермедиата
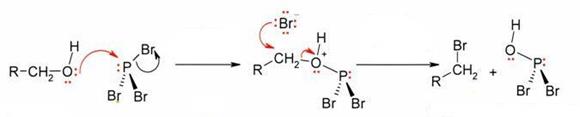
Для повышения выхода конечного продукта и уменьшения доли побочных реакций замещение ведут в присутствии пиридина.
В соответствии с особенностями механизма реакции (SN2), замещение гидроксильной группы на галоген происходит с обращением конфигурации у асимметрического атома углерода. При этом следует учитывать, что замещение часто осложняется изомеризацией и перегруппировками, поэтому подобная реакция, обычно, применяется для относительно спиртов простого строения.
Взаимодействие спиртов с тионилхлоридом
В зависимости от условий взаимодействие спиртов с SOCl2 протекает либо по механизму SNi, либо по механизму SN2. В обоих случаях спирт превращается в соответствующий алкилхлорид.
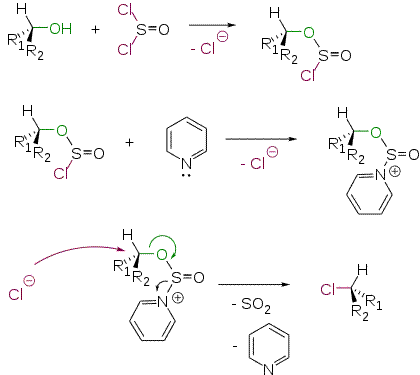
Если реакция проходит в отсутствие пиридина, продукт имеет такую же конфигурацию реакционного центра, что и исходный спирт (механизм SNi)
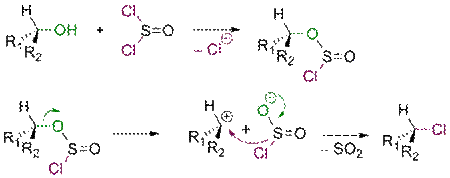
Добавление пиридина в реакционную смесь приводит к изменению стереохимического результата процесса. Полученный алкилхлорид имеет обращенную конфигурацию. Этот факт можно объяснить следующим механизмом SN2
Взаимодействие спиртов с хлорангидридами сульфокислот и последующим замещением
Спирты способны реагировать с хлорангидридами сульфокислот в присутствии основания с образованием соответствующих сложных эфиров. Первичные спирты реагируют быстрее вторичных и значительно быстрее третичных. Возможно селективное образование первичного сложного эфира сульфокислоты в присутствии вторичных и третичных спиртовых групп. Наибольшее практическое значение имеет получение алкилтозилатов (R−O−SO2C6H4CH3), алкилмезилатов (R−O−SO2CH3) и алкилтрифлатов (R−O−SO2CF3). В роли основания чаще всего используется пиридин, который одновременно выступает и как нуклеофильный катализатор. Сульфонаты являются прекрасными уходящими группами и легко замещаются на атом галогена по механизму SN2
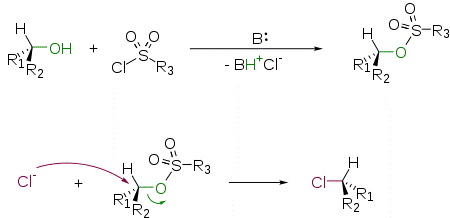
Источником галогенид-иона обычно является соответствующая неорганическая соль (NaBr, LiCl, CsF, KF] и т. д.) В качестве растворителя используются диполярные апротонные растворители: ДМСО, ДМФА, ацетонитрил. Замещение происходит, как правило, с обращением конфигурации.
Метод замещения гидроксила на высокореакционноспособную группу — мощный препаративный метод в органической химии, позволяющий получать из спиртов в две стадии, помимо галогенидов, самые различные соединения: простые эфиры, сложные эфиры карбоновых кислот, амиды.
Взаимодействие спиртов с квазифосфониевыми солями
Спирты могут быть превращены в алкилгалогениды реакцией с квазифосфониевыми солями — [R3PHal]+X−. Последние образуются при взаимодействии органофосфионов (R3P) с галогенами, тетрагалогенметанами (CCl4, CBr4) или N -галогенсукцинимидами (например, NBS). Данный метод применим к первичным и вторичным спиртам; в случае третичных спиртов возможно образование продуктов перегруппировки. R3PBr2 и R3PI2 (получаются из R3P и Br2/I2) дают хорошие выходы даже с третичными и неопентильными субстратами[96]. В общем виде реакция протекает по следующей схеме
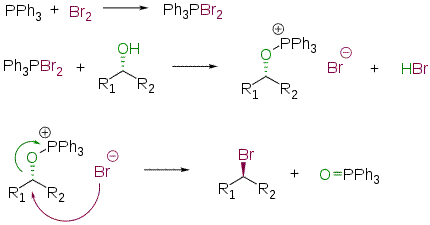
Превращение происходит с инверсией реакционного атома углерода.
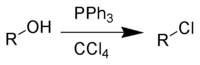
Частный случай взаимодействия — превращение спиртов в алкилхлориды под действием трифенилфосфина и тетрахлорметана — в заграничной литературе получил название реакции Аппеля (англ. Appel reaction)
Примеры неорганических реагентов, используемых для окисления спиртов
| Окислитель
| Исходное соединение
| Конечное соединение
| Условия реакции
|
| ацетат свинца(IV): Pb(CH3COO)4
| Ar−CH2OH
| Ar−CHO
| раствор в пиридине, комнатная температура
|
| R−CR'OH−CR'OH—R
| (RR')C=O
| уксуснокислый раствор, количественный выход
|
| тетраоксид диазота: N2O4
| R−CH2OH
| R−COOH
| хлороформ, 0 °С
|
| гипохлориты: Ca(OCl)2, NaOCl, KOCl
| R−CH2OH / R−CHOH−R
| R−C(O)−OCH2R / R−CO−R
| уксусная кислота, 0 °С
|
| нитрат диаммония-церия(IV): (NH4)2Ce(NO3)6
| Ar−CH2OH
| Ar−CHO
| уксусная кислота, 50—100 °С
|
| феррат калия: K2FeO4
| Ar−CH2OH + CH3OH
| Ar−COOCH3
| дихлорметан, CuSO4, выход более 70 %
|
| реагент Фетизона: Ag2CO3/кизельгур
| R−CH(OH)−R / R−CH(OH)-CH2-CH(OH)−R
| R−C(O)−R / R−C(O)-CH2-CH(OH)−R
| карбонат серебра, нанесённый на твёрдый носитель кизельгур (англ. celite)
|
Окисление с использованием активированного диметилсульфоксида. Окисление Пфицнера— Моффатта. В 1963 г. К. Пфицнером и Дж. Моффаттом была совершена публикация, в которой сообщалось об открытии нового метода окисления спиртов. Учёные растворяли исходные компоненты в смеси безводного диметилсульфоксида и дициклогексилкарбодиимида в присутствии слабой кислоты. В результате реакции в зависимости от строения спирта получался соответствующий альдегид или кетон, при этом даже для чувствительных первичных спиртов в продуктах окисления практически не 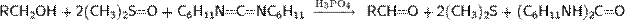 наблюдались следов карбоновых кислот
наблюдались следов карбоновых кислот
Спустя два года был предложен механизм превращения
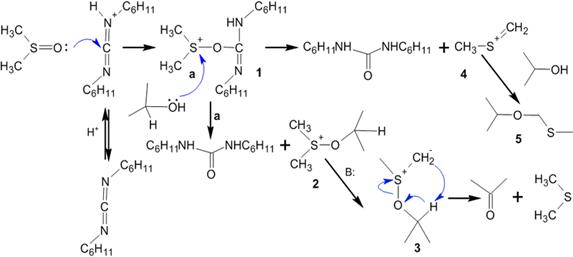
В соответствии с механизмом реакции протонированный дициклогексилкарбодиимид (ДЦК) на первом этапе вступает в реакцию с диметисульфоксидом (ДМСО) с образованием сульфониевого интермедиата (1), так называемого «активированного ДМСО», содержащего легко-уходящую группу, связанную с положительно заряженным атомом серы. Спирт быстро замещает эту группу, образуя алкоксидиметилсульфониевую соль (2), которая в свою очередь, теряя протон, превращается в тиоилид (3). В финальной стадии процесса происходит внутримолекулярное расщепление илида, проводящее к образованию конечного карбонильного соединения и диметилсульфида. Отмечается, что «Активированный ДМСО» (1) способен распадаться с образованием высокореакционной частицы (4), которая вступая в реакцию со спиртом, образует побочный продукт — метилтиометиловый эфир(5). Вместе с тем, учитывая, что элиминирование протекает при более высокой температуре, чем основной процесс, можно использовать температурный контроль хода реакции для минимизации доли побочных продуктов. Согласно механизму окисления, для протонирования ДЦК необходимо присутствие кислоты, однако сильные минеральные кислоты (HCl, HClO4, H2SO4 и т. п.) для реакции непригодны — они предотвращают образование илида (3). Проведённые эксперименты показали, что оптимальным является использование фосфорной или дихлоруксусной кислоты, а также трифторацетата пиридиния.
Данный метод стал основой для многочисленных научных исследований в области окисления спиртов активированным диметилсульфоксидом, что привело впоследствии к многочисленным модификациям и практическим разработкам новых способов окисления.
Окисление Олбрайта— Голдмана и Олбрайта— Онодера. В 1965 г. (спустя два года после сообщения Пфицнера и Моффатта) Олбрайтом и Голдманом был предложен способ окисления спиртов при комнатной температуре смесью ДМСО и уксусного ангидрида. Предложенная модификация уступает методу Пфицнера— Моффатта из-за большего количества побочных продуктов, однако доступность уксусного ангидрида делает окисление Олбрайта— Голдмана полезным для лабораторной практики.В том же сообщении 1965 г. Олбрайт и Голдман упомянули, что ДМСО можно активировать оксидом фосфора(V). Спустя несколько месяцев Онодера с сотрудниками сделал подробный доклад о новом методе окисления спиртов смесью ДМСО и P2O5 (метод получил название окисление Олбрайта— Онодера. Наконец, в 1987 г. данный способ окисления был улучшен: в качестве растворителя был использован дихлорметан в присутствии триэтиламина.
Окисление Париха— Деринга. Ещё одним методом окисления спиртов с использованием активированного диметилсульфоксида является окисление Париха— Деринга, где в качестве активирующего реагента используется раствор триоксида серы в пиридине (пиридиновый комплекс SO3•C5H5N) в присутствии триэтиламина. Реакция проходит при охлаждении (около 0 °С) или комнатной температуре. Метод, открытый Парихом и Дерингом в 1967 г., несмотря на его практическую доступность, отличается повышенным содержанием в целевых продуктах побочного компонента — метилтиометилового эфира. Механизм окисления Париха— Деринга аналогичен механизму окисления Пфицнера— Моффатта
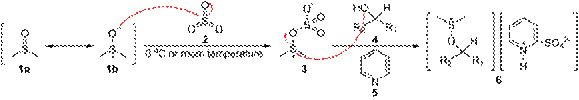
 Окисление Сверна. Одним из лучших методов, использующих активированный ДМСО, стал процесс с использованием оксалилхлорида, открытый в 1978 г. Сверном
Окисление Сверна. Одним из лучших методов, использующих активированный ДМСО, стал процесс с использованием оксалилхлорида, открытый в 1978 г. Сверном
Окисление спиртов по Сверну может быть выполнено в очень мягких условиях. С помощью этой реакции можно получать альдегиды и кетоны из первичных и вторичных спиртов соответственно. Главным недостатком метода является выделение токсичных и зловонных побочных продуктов— диметилсульфида и оксида углерода(II)
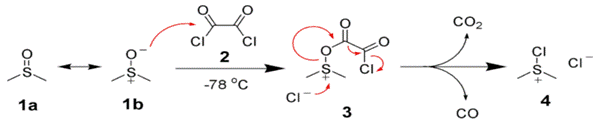
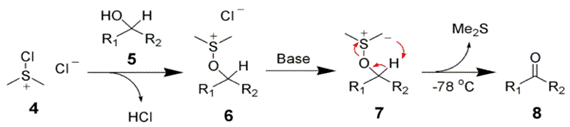
Первый этап реакции Сверна заключается в низкотемпературном взаимодействии диметилсульфоксида (1a и 1b) с оксалихлоридом (2). Промежуточный интермедиат (3) быстро разлагается с выделением CO и CO2 и образованием хлорида диметилхлорсульфония (4), который в свою очередь вступает в реакцию со спиртом (5), образуя ион алкоксисульфония (6). Далее в реакцию вступает триэтиламин, который депротонирует интермедиат, давая илид (7). Переходный пятичленный цикл (7) разлагается, образуя диметилсульфид и конечный кетон или альдегид (8).
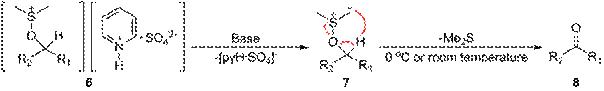
Окисление Кори— Кима. В отличие от окисления по Пфицнеру— Моффатту и ему подобных, где «активированный ДМСО» образуется в реакции ДМСО с электрофильным агентом, метод Кори— Кима использует в качестве исходного реагента диметилсульфид 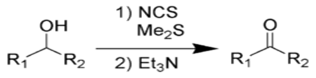
Сущность метода заключалась в образовании хлорида хлордиметилсульфония — представлявшего собой по сути «активированный ДМСО» Сверна— действием хлора на ДМС
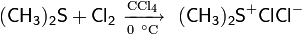
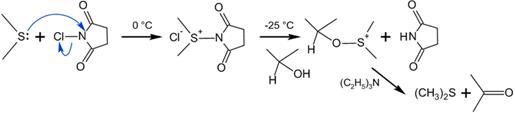
На практике, однако учёные предложили использовать вместо хлора N-хлор-сукцинимид (NCS), который вступая в реакцию с диметисульфидом, образует ион хлордиметилсульфония, а он в свою очередь реагирует со спиртом по аналогии с процессом Сверна
Окисление с использованием алкоголятов металлов.Окисление по Оппенауэру. В начале 20-го века независимо Меервейном, Пондорфом и Верлеем была открыта реакция восстановления карбонильных соединений в спирты (восстановление по Меервейну — Пондорфу — Верлею) в присутствии алкоголята алюминия (в качестве донора протонов выступал изопропанол) 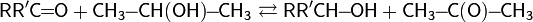
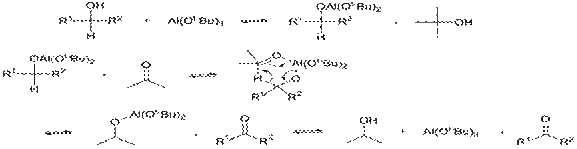 В 1937 г. Оппенауэром была осуществлена обратная реакция: используя в качестве окислителя избыток ацетона в присутствии трет -бутилата алюминия, ему удалось, по сути, сдвинуть равновесие и перенести процесс восстановления в обратную сторону
В 1937 г. Оппенауэром была осуществлена обратная реакция: используя в качестве окислителя избыток ацетона в присутствии трет -бутилата алюминия, ему удалось, по сути, сдвинуть равновесие и перенести процесс восстановления в обратную сторону
Окисление Мукаямы. В 1977 г. Мукаяма с сотрудниками опубликовал работу, в которой сообщал, что алкоголяты магния, образующиеся в результате взаимодействия спирта с пропилмагнийбромидом или трет -бутоксимагнийбромидом в присутствии
1,1’-(азодикарбонил)дипиперидина (выступает в роли акцептора водорода) при комнатной температуре окисляют исходный спирт до альдегида или кетона
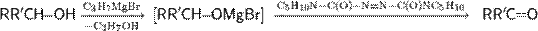
Хотя реакция Мукаямы и не принадлежит к числу распространённых методов окисления спиртов, она представляет препаративный интерес из-за более мягких условий протекания (по сравнению с окислением Оппенауэра) и сопровождается меньшим количеством побочных продуктов.
Прочие методы окисления. Окисление соединениями гипервалентного иода. Соединения пятивалентного иода — сильные окислители, однако из-за своей нестабильности и плохой растворимости в органических растворителях они практически не использовались в лабораторной органической практике. Однако, в 1983 г. Десс и Мартин опубликовали информацию о новом стабильном и хорошо растворимом в дихлорметане органическом соединении гипервалентного иода, являющегося эффективным и очень мягким окислителем для первичных и вторичных спиртов

Метод, получивший название окисления Десса— Мартина, оказался очень эффективным и получил свое развитие во многих последующих работах. Помимо периодинана Десса— Мартина существуют и другие соединения гипервалентного иода, используемые как окислители для спиртов: 2-иодоксибензойная кислота, дихлорид иодбензола, иодозобензол и др..
Окисление стабильными нитроксидными радикалами
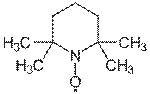
TEMPO — стабильный нитроксидный радикал. В 1987 г. Анелли с сотрудниками опубликовал исследование, в котором сообщалось об использовании свободного нитроксидного радикала (4-метокси-2,2,6,6-тетраметилпиперидин-1-оксил или англ. 4-метокси-TEMPO) в качестве катализатора для быстрого селективного окисления первичных и вторичных спиртов. Реакция проводилась при 0 °С в двухфазной среде CH2Cl2—вода в присутствии вторичного окислителя (NaOCl), а также небольших количеств NaHCO3 (стабилизирует pH раствора) и KBr (ускоряет реакцию вследствие образования HOBr — более сильного окислителя по сравнению с HOCl)
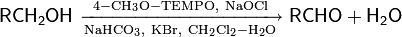
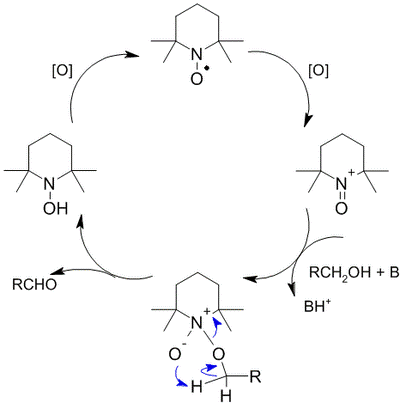
Механизм реакции окисления с использованием TEMPO выглядит следующим образом
В настоящий момен