Введение
Основу жизнедеятельности любого организма составляют химические процессы. Практически все реакции в живом организме протекают с участием природных биокатализаторов - ферментов.
Берцелиус в 1835 г. впервые предположил, что реакции живого организма осуществляются благодаря новой силе, которую он назвал «каталитической». Эту идею он обосновал главным образом экспериментальным наблюдением: диастаза из картофеля гидролизует крахмал быстрее, чем серная кислота. Уже в 1878 г. Куне назвал вещество, обладающее каталитической силой в живом организме, ферментом.
Кинетика действия ферментов - это раздел ферментологии, изучающий зависимость скорости реакции, катализируемой ферментами, от химической природы и условий взаимодействия субстрата с ферментом, а также от факторов среды. Иначе говоря, кинетика ферментов позволяет понять природу молекулярных механизмов действия факторов, влияющих на скорость ферментативного катализа. Этот раздел образовался на стыке таких наук, как биохимия, физика и математика. Самая ранняя попытка математически описать ферментативные реакции была предпринята Дюкло в 1898 г.
На самом деле этот раздел по изучению ферментов очень важен в наше время, а именно для практической медицины. Он даёт фармакологам инструмент направленного изменения метаболизма клетки, огромное количество фармацевтических препаратов и различные яды - это ингибиторы ферментов.
Целью данной работы является рассмотрение вопроса о зависимости скорости реакции от различных факторов, каким образом можно контролировать скорость реакций и как её можно определить.
Кинетика Михаэлиса - Ментен
Предварительные эксперименты по изучению кинетики ферментативных реакций показали, что скорость реакции 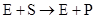 , вопреки теоретическим ожиданиям, не зависит от концентрации фермента (Е) и субстрата (S) таким образом, как в случае обычной реакции второго порядка.
, вопреки теоретическим ожиданиям, не зависит от концентрации фермента (Е) и субстрата (S) таким образом, как в случае обычной реакции второго порядка.
Браун и независимо от него Анри впервые выдвинули гипотезу об образовании в ходе реакции фермент-субстратного комплекса. Затем это предположение подтвердили три экспериментальных факта:
а) папаин образовывал нерастворимое соединение с фибрином (Вюртц, 1880);
б) субстрат инвертазы сахароза могла защищать фермент от тепловой денатурации (О'Салливан и Томпсон, 1890);
в) было показано, что ферменты являются стереохимически специфическими катализаторами (Фишер, 1898-1899).
В 1913 г. Михаэлис и Ментен опубликовали свою теорию общего механизма ферментативных реакций:

Они ввели понятие максимальной скорости и показали, что кривая насыщения (т.е. зависимость скорости реакции от концентрации субстрата) является равнобочной гиперболой. Они доказали, что максимально наблюдаемая скорость есть одна из асимптот к кривой, а отрезок, отсекаемый на оси абсцисс (в области ее отрицательных значений) второй асимптотой, т.е. константа в уравнении скорости, равен по абсолютному значению концентрации субстрата, необходимой для достижения половины максимальной скорости. [2]
Михаэлис и Ментен предположили, что скорость реакции определяется распадом комплекса ES, т.е. константой k2. Это возможно только при условии, что k2- наименьшая из констант скорости. В этом случае равновесие между фермент-субстратным комплексом, свободным ферментом и субстратом устанавливается быстро по сравнению со скоростью реакции (быстро устанавливающееся равновесие).
Начальную скорость реакции можно выразить следующей формулой:
v = k2 [ES]
Поскольку константа диссоциации фермент-субстратного комплекса равна
KS = [E] [S] / [ES] = k -1/k1
то концентрацию свободного фермента можно выразить как
[E] =KS [ES] / [S]
Общая концентрация фермента в реакционной смеси определяется формулой
[Е]т = [Е] + [ЕS] = KS [ЕS] / [S] + [ЕS]
Реакция достигает максимальной скорости, когда концентрация субстрата достаточно высока, чтобы все молекулы фермента находились в виде комплекса ЕS (бесконечно большой избыток субстрата). Отношение начальной скорости к теоретически возможной максимальной скорости равно отношению [ЕS] к [Е]т:
v / Vmax= [ES] / [E]т= [ES] / (KS [ES] / [S] + [ES]) = 1 / (KS+[S] +1)

Это классическое уравнение Михаэлиса и Ментен, которое со времени его публикации в 1913 г. стало фундаментальным принципом всех кинетических исследований ферментов в течение десятилетий и с некоторыми ограничениями осталось таким до сих пор. [5]
Позднее было показано, что оригинальное уравнение Михаэлиса - Ментен предполагало наличие нескольких ограничений. Оно справедливо, т.е. правильно описывает кинетику реакции, катализируемой данным ферментом, только при условии выполнения всех следующих ограничительных условий:
) образуется кинетически устойчивый фермент-субстратный комплекс;
) константа KSявляется константой диссоциации фермент-субстратного комплекса: это справедливо, только если  ;
;
) концентрация субстрата не меняется в ходе реакции, т.е. концентрация свободного субстрата равна его начальной концентрации;
) продукт реакции быстро отщепляется от фермента, т.е. не образуется кинетически значимого количества ЕS комплекса;
) вторая стадия реакции необратима; точнее говоря, мы принимаем во внимание только начальную скорость, когда обратной реакцией (из-за фактического отсутствия продукта) еще можно пренебречь;
) с каждым активным центром фермента связывается только одна молекула субстрата;
) для всех реагирующих веществ вместо активностей можно использовать их концентрации. [2]
Уравнение Михаэлиса - Ментен служит отправной точкой при любом количественном описании действия ферментов. Следует подчеркнуть, что кинетическое поведение большинства ферментов значительно сложнее, чем это вытекает из идеализированной схемы, лежащей в основе уравнения Михаэлиса - Ментен. При выводе этого уравнения предполагается, что существует только один фермент-субстратный комплекс. Между тем в действительности в большинстве ферментативных реакций образуется, по меньшей мере, два или три таких комплекса, возникающих в определенной последовательности.
Здесь через EZ обозначен комплекс, соответствующий истинному переходному состоянию, а через ЕР - комплекс между ферментом и продуктом реакции. Можно указать также, что в большинстве ферментативных реакций участвует более одного субстрата и образуется соответственно два или большее число продуктов. В реакции с двумя субстратами, S1 и S2, может образоваться три фермент-субстратных комплекса, а именно ES1, ES2 и ES1S2. Если в результате реакции получается два продукта, P1 и P2, то может существовать, по меньшей мере, еще три дополнительных комплекса EP1, EP2 и EP1P2. В таких реакциях имеется много промежуточных стадий, каждая из которых характеризуется своей константой скорости. Кинетический анализ ферментативных реакций, в которых принимают участие два реагирующих вещества или более, часто оказывается исключительно сложным и требует использования электронных вычислительных машин. Тем не менее, при анализе кинетики всех ферментативных реакций отправной точкой всегда является рассмотренное выше уравнение Михаэлиса - Ментен. [5]
Образование кинетически устойчивого комплекса фермент - продукт
Если в ходе реакции происходит образование кинетически устойчивого комплекса фермент - продукт, механизм реакции выглядит следующим образом:

Применив предположение о стационарном состоянии, можно написать дифференциальные уравнения:
d [ES] /dt = k1 [E] [S] + k-2 [EP] - (k-1 + k2) [ES] = 0[EP] /dt = k2 [ES] - (k-2 + k3) [EP] = 0
Из этих уравнений следует, что
[ES] = [(k-2 + k3) / k2] [EP]
[E] = [(k-1 k-2 + k-1 k-3 + k2k3) / k1k2 [S]] [EP]
Так как v = k3 [EP]
и [E]T = [E] + [ES] + [EP] =
= [(k-1 k-2 + k-1 k-3 + k2k3) / k1k2 [S] + (k-2 + k3) / k2 + 1] [EP] =
= {[k-1 k-2 + k-1 k-3 + k2k3 + k1 [S] (k-2 + k3) + k1k2 [S]] / k1k2 [S]} [EP]
получаем
[EP] = k1k2[S] [E]T / [k-1 k-2 + k-1 k-3 + k2k3 + k1 [S] (k-2 + k3 + k2)]= k1k2k3[S] [E]T / [k-1 k-2 + k-1 k-3 + k2k3 + k1 [S] (k-2 + k3 + k2)] =
= [k2k3 / (k-2 + k3 + k2)] [E]T[S] / [(k-1 k-2 + k-1 k-3 + k2k3) / k1 (k-2 + k3 + k2) + [S]]
То есть
Vmax = [k2k3 / (k-2 + k3 + k2)] [E]Tm = (k-1 k-2 + k-1 k-3 + k2k3) / k1 (k-2 + k3 + k2)
В этом случае уже очень сложно вычислить конкретные значения индивидуальных констант скорости, так как прямо измерить можно только их отношение. Ситуация еще более затрудняется при усложнении механизма ферментативной реакции, когда в реакции участвуют больше двух комплексов, потому что количество констант скорости в уравнении, естественно, гораздо больше, и их соотношения также сложнее. [2]
Однако ситуация упрощается, если после обратимой реакции образования первого комплекса последующие элементарные стадии необратимы. Важными представителями ферментов, подчиняющихся этому механизму, являются протеолитические ферменты и эстеразы. Механизм их реакции можно записать следующим образом:
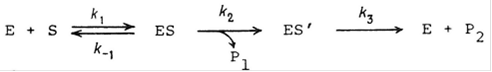
где ES` - ацилферментное промежуточное соединение, которое разлагается под действием воды. Мы можем написать
d [P2] /dt = d [P1] / dt = v = k1k2k3 [S] [E]0 / [k3(k-1 + k2) + [S] (k2 + k3)]
Vmax = k2k3 [E]0 / (k2 + k3) = kкат [E]0m = k3 (k-1 + k2) / (k2 + k3) k1кат / Km = k2k1 / (k-1 + k2) = k2 / Km’
Константа Михаэлиса стадии ацилирования - Km'  Ks. Чем больше отношение kкат/Km, тем выше специфичность субстрата. [5]
Ks. Чем больше отношение kкат/Km, тем выше специфичность субстрата. [5]
Определение констант значительно упрощается, если эксперимент проводят в присутствии нуклеофильного агента (N), способного конкурировать с водой. Тогда

k3 = k3’ [H2O] и Pi (i = 1, 2, 3) - продукты.
vi = kкат, i [E0] [S] / (Km + [S])кат, 1 = k2 (k3 + k4 [N]) / (k2 + k3 + k4 [N])кат, 2 = k2k3 / (k2 + k3 + k4 [N])кат, 3 = k2k4 [N] / (k2 + k3 + k4 [N])m = Ks (k3 + k4 [N]) / (k2 + k3 + k4 [N])
/vN = Ks (k3 + k4 [N]) / k2k3 [S] [E0] + (k2 + k3 + k4 [N]) / k2k3 [E0]
Так как известно, что Ks/k2 = Km/ kкат, и если нуклеофил отсутствует, то
1/v = Ks / k2 [S] [E0] + (k2 + k3) / k2k3 [E0]
и для определения констант можно использовать точку пересечения прямых в координатах 1/vN (и 1/v) - 1/[S]. Две прямые линии в двойных обратных координатах пересекаются во втором квадранте. В отсутствии нуклеофила точка пересечения прямой с вертикальной осью определяется как 1/Vmax и 1/kкат[E0], а с горизонтальной осью - как -1/Km. Координаты точки пересечения двух прямых: -1/Ks и 1/k3[E0]. Расстояние между 1/Vmax и 1/k3[E0] равно 1/k2[E0].
Активация ферментов
Регуляция ферментов может осуществляться путем взаимодействия с ними различных биологических компонентов или чужеродных соединений (например, лекарств и ядов), которые принято называть модификаторами или регуляторами ферментов. Под действием модификаторов на фермент реакция может ускоряться (активаторы) или замедляться ( ингибиторы).
Активация ферментов определяется по ускорению биохимических реакций, наступающему после действия модификатора. Одну группу активаторов составляют вещества, влияющие на область активного центра фермента. К ним относятся кофакторы ферментов и субстраты. Кофакторы (ионы металлов и коферменты) являются не только обязательными структурными элементами сложных ферментов, но и по существу их активаторами.
Ионы металлов бывают довольно специфичными активаторами. Часто для некоторых ферментов требуются ионы не одного, а нескольких металлов. Например, для Na+, K+-АТФазы, осуществляющей транспорт одновалентных катионов через клеточную мембрану, необходимы в качестве активаторов ионы магния, натрия и калия.
Активация с помощью ионов металлов осуществляется по разным механизмам. В некоторых ферментах они входят в состав каталитического участка. В ряде случаев ионы металлов облегчают связывание субстрата с активным центром фермента, образуя как бы своеобразный мостик. Нередко металл соединяется не с ферментом, а с субстратом, образуя металлосубстратный комплекс, который предпочтителен для действия фермента. [1]
Специфичностью участия коферментов в связывании и катализе субстрата объясняется активация ими ферментативных реакций. Особенно заметно активирующее влияние кофакторов при действии на фермент, который не насыщен кофакторами.
Субстрат тоже в известных пределах концентраций является активатором. После достижения насыщающих концентраций субстрата активность фермента не возрастает. Субстрат повышает стабильность фермента и облегчает формирование нужной конформации активного центра фермента.
Ионы металлов, коферменты и их предшественники и активные аналоги,
субстраты можно использовать на практике как препараты, активирующие ферменты.
Активация некоторых ферментов может осуществляться путем модификации, не затрагивающей активный центр их молекул. Возможно несколько вариантов такой модификации:
1) активация неактивного предшественника - профермента, или зимогена. Например, превращение пепсиногена в пепсин;
2) активация путем присоединения какой-либо специфической модифицирующей группы к молекуле фермента;
3) активация путем диссоциации неактивного комплекса белок - активный фермент. [6]
Ингибирование ферментов
Существуют реагенты, способные взаимодействовать более или менее специфично с той или иной боковой цепью белков, что приводит к ингибированию активности фермента. Это явление позволяет изучать природу аминокислотных боковых остатков, принимающих участие в данной ферментативной реакции. Однако на практике следует учитывать многочисленные тонкости, делающие однозначную интерпретацию результатов, полученных со специфическими ингибиторами, довольно трудной и зачастую сомнительной. Прежде всего, чтобы реакция с ингибитором подходила для изучения природы участвующих в реакции боковых цепей, она должна удовлетворять следующим критериям:
) быть специфичной, т.е. ингибитор должен блокировать только нужные группы;
) ингибировать активность фермента, и это ингибирование должно становиться полным при увеличении числа модифицированных групп;
) реагент не должен вызывать неспецифическую денатурацию белка.
Выделяют 2 группы ингибиторов: обратимого и необратимого действия. В основе подразделения лежит критерий восстановления активности фермента после диализа или сильного разведения раствора фермента с ингибитором.
По механизму действия выделяют конкурентное, неконкурентное, бесконкурентное, субстратное и аллостерическое ингибирование. [3]
Конкурентное ингибирование
Конкурентное ингибирование было открыто при изучении ингибирования, вызываемого аналогами субстрата. Это торможение ферментативной реакции, вызванное связыванием с активным центром фермента ингибитора сходного по структуре с субстратом и препятствующего образованию фермент-субстратного комплекса. При конкурентном торможении ингибитор и субстрат, будучи сходными по строению, конкурируют за активный центр фермента. С активным центром связывается то соединение молекул, которого больше.
Такие представления о механизме ингибирования были подтверждены экспериментами по кинетике реакций конкурентного ингибирования. Так, было показано, что в случае конкурентного ингибирования аналог субстрата не влияет на скорость разложения уже образовавшегося комплекса фермент-субстрат, т.е. при использовании «бесконечно большого» избытка субстрата получается одна и та же максимальная скорость как в присутствии, так и в отсутствие ингибитора. Напротив, ингибитор влияет на величину константы диссоциации и константы Михаэлиса. Из этого можно сделать вывод, что ингибитор реагирует с группами белка, участвующими тем или иным образом в связывании субстрата, следовательно, из-за взаимодействия его с этими группами прочность связывания субстрата уменьшается (т.е. уменьшается число молекул фермента, способных связывать субстрат).
Позже было показано, что кинетически конкурентное ингибирование может быть вызвано не только аналогами субстратов, но и другими реагентами, химическая структура которых абсолютно отличается от структуры субстрата. В этих случаях также предполагалось, что данный реагент взаимодействует с группой, ответственной за связывание субстрата.
Для конкурентного ингибирования теоретически могут существовать две возможности:
1)связывающие и каталитические центры фермента перекрываются; ингибитор связывается с ними, но влияет только на группы центра связывания;
2)центр связывания и каталитический центр в молекуле фермента пространственно обособлены; ингибитор взаимодействует с центром связывания.
Существуют следующие элементарные стадии реакции:

где I - ингибитор, а KI - константа диссоциации комплекса фермент - ингибитор.
Относительная скорость (отношение скорости ферментативной реакции, измеренной в присутствии ингибитора (vi), к максимальной скорости) равна
vi / V = [ES] / [E]T
поскольку для общей концентрации фермента справедливо
[E]T = [E] + [ES] + [EI]
то 1 / vi = (Ks / V[S]) (1 + [I] / KI) + 1 / V
Очевидно, если [I] = KI, то наклон прямой линии становится вдвое больше, чем для зависимости 1/v0 от [S] (v0 - скорость ферментативной реакции в отсутствие ингибитора).
Тип ингибирования обычно определяют графически. Конкурентное ингибирование легче всего распознается путем построения графиков Лайнуивера - Берка (т.е. графиков в координатах 1/viи 1/[S]) при разных концентрациях ингибитора. При истинном конкурентном ингибировании получается набор прямых, отличающихся тангенсом угла наклона и пересекающих ось ординат (ось 1/vi)в одной точке. При любой концентрации ингибитора можно попользовать настолько высокую концентрацию субстрата, что активность фермента будет максимальной.
В качестве примера конкурентного ингибирования можно привести влияние различных веществ на активность сукцинатдегидрогеназы. Этот фермент входит в состав ферментной циклической системы - цикла Кребса. Его природным субстратом является сукцинат, а сходным с ним конкурентным ингибитором - оксалоацетат, промежуточный продукт того же цикла Кребса:

Аналогичным конкурентным ингибитором сукцинатдегидрогеназы является малоновая кислота, часто использующаяся в биохимических исследованиях.
На принципе конкурентного ингибирования основано действие многих фармакологических препаратов, ядохимикатов, используемых для уничтожения сельскохозяйственных вредителей, и боевых отравляющих веществ.
Например, группа антихолинэстеразных препаратов, к которым относятся производные четвертичных аммониевых оснований и фосфорорганические соединения, являются конкурентными ингибиторами фермента холинэстеразы по отношению к его субстрату ацетилхолину. Холинэстераза катализирует гидролиз ацетилхолина - медиатора холинэргических систем (нервно-мышечных синапсов, парасимпатической системы и т.д.). Антихолинэстеразные вещества конкурируют с ацетилхолином за активный центр фермента, связываются с ним и выключают каталитическую активность фермента. Такие препараты, как прозерин, физостигмин, севин, угнетают фермент обратимо, а фосфорорганические препараты типа армина, нибуфина, хлорофоса, зомана действуют необратимо, фосфорилируя каталитическую группу фермента. В результате их действия накапливается ацетилхолин в тех синапсах, где он является медиатором нервного возбуждения, т.е. происходит отравление организма накопившимся ацетилхолином. Действие обратимых ингибиторов постепенно проходит, так как чем больше накапливается ацетилхолина, тем быстрее он вытесняет ингибитор из активного центра холинэстеразы. Токсичность необратимых ингибиторов несравненно выше, поэтому их применяют для борьбы с вредителями сельского хозяйства, бытовыми насекомыми и грызунами (например, хлорофос) и как боевые отравляющие вещества (например, зарин, зоман и др.). [6]
Субстратное ингибирование
Субстратным ингибированием называется торможение ферментативной реакции, вызванное избытком субстрата. Такое ингибирование происходит вследствие образования фермент-субстратного комплекса, не способного подвергаться каталитическим превращениям, Комплекс ES2 непродуктивный и делает молекулу фермента неактивной. Субстратное торможение вызвано избытком субстрата, поэтому снимается при снижении его концентрации. [6]
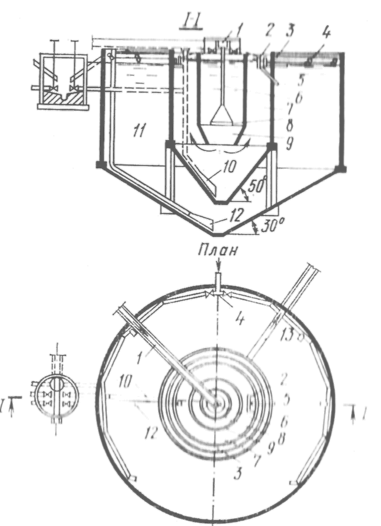
Устройство к рН-метру
Устройство к рН-метру состоит из модифицированного наконечника проточного электрода 1, полумикроячейки 2, дозатора 3 и электронной схемы подключения рН-метра к самописцу. Кроме того, устройство включает стандартный электрод рН-метра (4), крышку-держатель ячейки (5), термостатическую проточную камеру (6), раствор субстрата (7), пассивный магнит (8), активный магнит (9).
Стандартный наконечник проточного электрода рН-метра (ЛПУ-01) заменяется тефлоновой трубкой 1 (внутренний диаметр 1,3-1,5 мм), заполняется асбестовой нитью, предварительно обработанной насыщенным раствором KCl. Плотность заполнения нити регулируется таким образом, чтобы скорость протока раствора KCl через трубку была близкой к скорости протока исходного немодифицированного электрода. Такая замена наконечника дает возможность снизить размеры исходной рабочей ячейки с 20-25 до 2 мл, что позволяет обходиться минимальными объемами (1,5 мл) растворов дорогостоящих биохимических препаратов. [4]
Электронная схема подключения рН-метра (ЛПУ-01) к самописцу состоит из источника питания (батареи постоянного тока 12 В), переменного проволочного сопротивления R1 (10 - 100 Ом), задающего по показанию вольтметра напряжение 9 В на стабилотроне Д809, переменного проволочного сопротивления R2 (15-150 Ом), регулирующего установку «нуля» (начала отсчета) показаний рН-метра на шкале самописца, и переменного проволочного сопротивления R3 (35-500 Ом), регулирующего масштаб расширения (усиления) показаний шкалы рН-метра на самописце. Схема работает надежно до падения напряжения источника не ниже 9 В.
Принцип работы. В ячейку (стеклянный цилиндр 1,7х2,4 см) вносится 1,5 мл субстрата, и ячейка закрепляется на крышке-фиксаторе 5. Включается перемешивание 9, и перо самописца пишет ровную (базисную) линию отсчета. При помощи дозатора 0,03 мл раствора фермента вносится в субстрат, и перо самописца фиксирует начало реакции отклонением кривой зависимости рН от времени (t).
Такое устройство не заменяет рН-стата, но с учетом возможности расширения шкалы рН-метра позволяет надежно фиксировать незначительные изменения рН 0,004-0,005.
3.3 Номограммные линейки, удобные для определения начальной скорости
Значительную трудоёмкость определений начальной скорости в методе касательных составляет подсчёт отношений изменения концентраций реагентов (Δ[S]) за единицу времени (Δt), т.е. выражение v0 в М/мин из условий, что
v0 = lim Δ[S] / Δt, при, t 0.
На практике такая процедура складывается обычно из трех-четырех отдельных операций: проводят касательную к начальному участку кривой хода реакции, затем подсчитывают число единиц регистрируемой величины (оптическая плотность, угол вращения и т.п.), приходящихся на определенный интервал времени, приводят это к единице времени и, наконец, делают пересчет показаний самописца на изменение концентраций реагента за 1 мин (М/мин). Предлагаемые два типа номограммной линейки позволяют упростить эту процедуру.
Прямоугольная линейка. v0 есть отношение Δ[S]/Δt, т.е. tg ά, где ά - угол наклона касательной к оси времени t. Эта же касательная является и гипотенузой соответствующего прямоугольного треугольника с катетами [S] иt. Чем больше v0, тем круче наклон касательной. Следовательно, если мы ограничимся определенным интервалом времени, например 1 мин, то получим серию прямоугольных треугольников с разной величиной катета [S] (в действительности разной величиной v0). Если же проградуировать оба катета: горизонтальный - в единицах отсчета времени (1 мин), а вертикальный- в единицах изменения концентраций реагента, например в миллимолях (мМ), и нанести полученные отрезки на подходящий формат из прозрачного материала (оргстекло толщиной около 2 мм), то можно получить удобную линейку для определения начальных скоростей реакций. Все цифры и линии наносятся на обратной стороне линейки, чтобы исключить погрешности на параллакс при определениях v0.
Процедура определения v0 сокращается в этом случае до двух простых операций: к начальному участку кинетической кривой t проводят касательную 2 и совмещают нулевую точку горизонтального катета t линейки с началом касательной, продолжение касательной пересечет теперь шкалу концентраций [S] в точке, определяющей значение v0 в М/мин (при горизонтальном положении катета t на. Никаких дополнительных операций больше не требуется. [4]
Дуговая линейка. Процедуру определения v0 можно упростить до одной операции, если шкалу концентраций отложить по дуге определенного радиуса.
На пластинку из прозрачного материала наносят прямую («базисную») линию 2 (все цифры и линии также наносят на обратной стороне линейки) и из нулевой точки (t=0, мин) этой линии радиусом, равным длине катета t=1 мин [, проводят дугу [S], сверху вниз по которой откладывают шкалу изменения концентраций реагента (например, субстрата в мМ).
Процедура определения v0 сводится в этом случае к одной операции. На кинетическую кривую 1 накладывают линейку так, чтобы ее «базисная» линия 2 к начальному участку кривой 1, а нулевая точка (0) этой базисной линии находилась на одной из горизонтальных линий 3 бумаги самописца. Продолжение этой горизонтальной линии в таком случае пересечет шкалу концентраций расщепляемого субстрата (дуга [S]) в точке, определяющей значение v0 в М/мин. Никаких дополнительных операций и в данном случае больше не требуется. [4]
Описанные типы линеек, устройство к спектрофотометру и рН-метру в течение ряда лет используются для определения начальных скоростей реакций (v0), при исследовании субстратной специфичности ферментов, для спектрофотометрического титрования и т.п.
Заключение
В данной работе был рассмотрен раздел энзимологии, изучающий зависимость скорости химических реакций, катализируемых ферментами, от ряда факторов окружающей среды. Основоположниками данной науки по праву считаются Михаэлис и Ментен, к оторые опубликовали свою теорию общего механизмаферментативных реакций, вывели уравнение, ставшее фундаментальным принципом всех кинетических исследований ферментов, оно служит отправной точкой при любом количественном описании действия ферментов. Исходное уравнение Михаэлиса - Ментен является уравнением гиперболы; свой вклад в кинетику внесли Лайнуивер и Бэрк, которые преобразовали уравнение Михаэлиса - Ментен и получили график прямой, по которой можно наиболее точно определить значение Vmax.
С течением времени изменение скорости ферментативной реакции в ферментативной реакции в экспериментальных условиях уменьшается. Снижение скорости может происходить за счёт ряда факторов: уменьшение концентрации субстрата, увеличение концентрации продукта, который может оказывать ингибирующее действие, могут происходить изменения рН раствора, изменения температуры среды. Так при повышении температуры на каждые 10°С скорость реакции увеличивается в 2 раза и даже меньше. Низкая температура обратимо инактивирует ферменты. Зависимость скорости ферментативной реакции от рН свидетельствует о состоянии функциональных групп активного центра фермента. Каждый фермент по-разному реагирует на изменение рН. Химические реакции можно останавливать путём действия на них различными видами ингибирования. Начальную скорость реакции можно быстро и точно определить при помощи таких приспособлений, как номограммные линейки, устройство к спектрофотометру и рН-метру. Это позволяет наиболее точно представить активность изучаемых ферментов.
Всё это активно используется в наши дни в медицинской практике.
Введение
Основу жизнедеятельности любого организма составляют химические процессы. Практически все реакции в живом организме протекают с участием природных биокатализаторов - ферментов.
Берцелиус в 1835 г. впервые предположил, что реакции живого организма осуществляются благодаря новой силе, которую он назвал «каталитической». Эту идею он обосновал главным образом экспериментальным наблюдением: диастаза из картофеля гидролизует крахмал быстрее, чем серная кислота. Уже в 1878 г. Куне назвал вещество, обладающее каталитической силой в живом организме, ферментом.
Кинетика действия ферментов - это раздел ферментологии, изучающий зависимость скорости реакции, катализируемой ферментами, от химической природы и условий взаимодействия субстрата с ферментом, а также от факторов среды. Иначе говоря, кинетика ферментов позволяет понять природу молекулярных механизмов действия факторов, влияющих на скорость ферментативного катализа. Этот раздел образовался на стыке таких наук, как биохимия, физика и математика. Самая ранняя попытка математически описать ферментативные реакции была предпринята Дюкло в 1898 г.
На самом деле этот раздел по изучению ферментов очень важен в наше время, а именно для практической медицины. Он даёт фармакологам инструмент направленного изменения метаболизма клетки, огромное количество фармацевтических препаратов и различные яды - это ингибиторы ферментов.
Целью данной работы является рассмотрение вопроса о зависимости скорости реакции от различных факторов, каким образом можно контролировать скорость реакций и как её можно определить.
Кинетика Михаэлиса - Ментен
Предварительные эксперименты по изучению кинетики ферментативных реакций показали, что скорость реакции , вопреки теоретическим ожиданиям, не зависит от концентрации фермента (Е) и субстрата (S) таким образом, как в случае обычной реакции второго порядка.
Браун и независимо от него Анри впервые выдвинули гипотезу об образовании в ходе реакции фермент-субстратного комплекса. Затем это предположение подтвердили три экспериментальных факта:
а) папаин образовывал нерастворимое соединение с фибрином (Вюртц, 1880);
б) субстрат инвертазы сахароза могла защищать фермент от тепловой денатурации (О'Салливан и Томпсон, 1890);
в) было показано, что ферменты являются стереохимически специфическими катализаторами (Фишер, 1898-1899).
В 1913 г. Михаэлис и Ментен опубликовали свою теорию общего механизма ферментативных реакций:
Они ввели понятие максимальной скорости и показали, что кривая насыщения (т.е. зависимость скорости реакции от концентрации субстрата) является равнобочной гиперболой. Они доказали, что максимально наблюдаемая скорость есть одна из асимптот к кривой, а отрезок, отсекаемый на оси абсцисс (в области ее отрицательных значений) второй асимптотой, т.е. константа в уравнении скорости, равен по абсолютному значению концентрации субстрата, необходимой для достижения половины максимальной скорости. [2]
Михаэлис и Ментен предположили, что скорость реакции определяется распадом комплекса ES, т.е. константой k2. Это возможно только при условии, что k2- наименьшая из констант скорости. В этом случае равновесие между фермент-субстратным комплексом, свободным ферментом и субстратом устанавливается быстро по сравнению со скоростью реакции (быстро устанавливающееся равновесие).
Начальную скорость реакции можно выразить следующей формулой:
v = k2 [ES]
Поскольку константа диссоциации фермент-субстратного комплекса равна
KS = [E] [S] / [ES] = k -1/k1
то концентрацию свободного фермента можно выразить как
[E] =KS [ES] / [S]
Общая концентрация фермента в реакционной смеси определяется формулой
[Е]т = [Е] + [ЕS] = KS [ЕS] / [S] + [ЕS]
Реакция достигает максимальной скорости, когда концентрация субстрата достаточно высока, чтобы все молекулы фермента находились в виде комплекса ЕS (бесконечно большой избыток субстрата). Отношение начальной скорости к теоретически возможной максимальной скорости равно отношению [ЕS] к [Е]т:
v / Vmax= [ES] / [E]т= [ES] / (KS [ES] / [S] + [ES]) = 1 / (KS+[S] +1)
Это классическое уравнение Михаэлиса и Ментен, которое со времени его публикации в 1913 г. стало фундаментальным принципом всех кинетических исследований ферментов в течение десятилетий и с некоторыми ограничениями осталось таким до сих пор. [5]
Позднее было показано, что оригинальное уравнение Михаэлиса - Ментен предполагало наличие нескольких ограничений. Оно справедливо, т.е. правильно описывает кинетику реакции, катализируемой данным ферментом, только при условии выполнения всех следующих ограничительных условий:
) образуется кинетически устойчивый фермент-субстратный комплекс;
) константа KSявляется константой диссоциации фермент-субстратного комплекса: это справедливо, только если ;
) концентрация субстрата не меняется в ходе реакции, т.е. концентрация свободного субстрата равна его начальной концентрации;
) продукт реакции быстро отщепляется от фермента, т.е. не образуется кинетически значимого количества ЕS комплекса;
) вторая стадия реакции необратима; точнее говоря, мы принимаем во внимание только начальную скорость, когда обратной реакцией (из-за факти