

Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...

Архитектура электронного правительства: Единая архитектура – это методологический подход при создании системы управления государства, который строится...
Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Архитектура электронного правительства: Единая архитектура – это методологический подход при создании системы управления государства, который строится...
Топ:
Выпускная квалификационная работа: Основная часть ВКР, как правило, состоит из двух-трех глав, каждая из которых, в свою очередь...
Процедура выполнения команд. Рабочий цикл процессора: Функционирование процессора в основном состоит из повторяющихся рабочих циклов, каждый из которых соответствует...
Интересное:
Как мы говорим и как мы слушаем: общение можно сравнить с огромным зонтиком, под которым скрыто все...
Берегоукрепление оползневых склонов: На прибрежных склонах основной причиной развития оползневых процессов является подмыв водами рек естественных склонов...
Искусственное повышение поверхности территории: Варианты искусственного повышения поверхности территории необходимо выбирать на основе анализа следующих характеристик защищаемой территории...
Дисциплины:
|
из
5.00
|
Заказать работу |
Введение
Создание и совершенствование компьютерной техники, появление новых технологий передачи, обработки, накопления и представления информации облегчило и ускорило вычислительную работу во многих областях науки и, в частности, химии. Квантовая химия, молекулярная механика, планирование химического синтеза, получение и обработка экспериментальных данных с помощью новых информационных технологий и компьютерной техники - это лишь некоторые типичные примеры. Увеличение вклада информационных в развитие химии связано в первую очередь с появлением персональных компьютеров с высокими эксплуатационными характеристиками и новых технологий передачи информации. Благодаря их относительно низкой цене и доступности для широкого круга пользователей такое оборудование стало обычным в большинстве химических лабораторий и учебных заведениях мира.
Сферами использования компьютеров среди химиков является подготовка текстов, содержащих химические структуры, визуализация пространственной структуры молекул, прогнозирование физико-химических свойств органических соединений на основании их химического строения и т.д. Популярные текстовые редакторы, такие как Microsoft Word, Word Perfect или WordPro и др., несмотря на свои широкие возможности, не имеют удобных инструментов для создания химических структур высокого качества. Для решения задач создания химических структур, схем реакций в полиграфических целях, использования их при создании баз данных, поиска в информационных системах, программах квантовой химии и других областях создано много различных программных средств. Одной из них является - ChemOffice.
Интегрированный программный комплекс ChemOffice включает следующие четыре специализированных приложения:
1) «химический редактор» CS Сhem Draw, являющийся традиционным средством редактирования химических формул;
2) специальный редактор баз данных СS СhemFinder, предназначенный для создания, редактирования и управления базами данных химических соединений;
) программа CS Chem3D, предназначенная для визуализации химических соединений, компьютерного моделирования и расчётов;
) редактор таблиц CS Table Editor, предназначенный для просмотра и редактирования табличных данных, используемых в пакете CS Chem3D.
Одним из важнейших элементов химического исследования является анализ геометрической структуры соединений. Эта область науки получила название структурная химия. Важнейшими экспериментальными методами, позволяющими исследовать геометрическую структуру молекул, является адсорбционная и эмиссионная спектроскопия. А также дифракционные методы. Структурные формулы отражают связанность различных атомов и молекул друг с другом. Классическим примером двухмерной структурной модели является структурная формула бензола: Брутто-формула С6Н6 не позволяет передать взаимосвязь между атомами углерода кольца, в связи с чем возникает необходимость отображения структуры в виде геометрического образца - задача визуализации структуры. Ещё более актуальной эта задача становиться при изучении стереоизомеров. Конформационный анализ соединений требует использование трёхмерной модели молекул. До появления компьютеров в качестве таких моделей широко применялись механические модели молекул (модели Стьюарта-Бриглеба). За последние десятилетие создано большое количество различных программных пакетов, позволяющих решать задачи визуализации как плоских, так и пространственных моделей молекул. Первые версии программ позволяли решать в основном задачи пространственных структур. В дальнейшим круг задач, решаемых с использованием специализированных «химических» программных пакетов, был расширен, и в настоящее время наиболее мощные из них реализуют практически все основные задачи компьютерной химии, включая управление базами данных химических соединений, методы квантовой химии и численного моделирования.
Целью данной работы было изучить возможности программного комплекса СhemOffice для построения, корректирования и двухмерной и трёхмерной визуализации химических формул.
В связи с поставленной целью нами решались следующие задачи:
1) Описать программное приложение ChemDraw 8.0 и выявить его возможности для двумерного построения и редактирования структурных химических формул;
2) Описать программное приложение Сhem3D Ultra 8.0 и определить его возможности для трёхмерной визуализации химических формул;
3) Определить различные способы редактирования и анализа геометрии трехмерных моделей молекул.
Главное меню
Как и в любом стандартном приложении Windows, Chem3D Ultra 6.0. имеет главное меню, расположенное вверху экрана. Посредством его осуществляется доступ к основным функциям пакета.
Пункт меню File (Файл) содержит стандартный набор возможностей для открытия, сохранения и печати файлов.
· Пункт New Model (Новая модель) открывает новое окно пользователя. С, что Chem3D Ultra 8.0 работает в многооконном режиме.
· Пункт Templates (Заготовки) предлагает открыть новое окно с уже заданными настройками (например, цвет фона или способ отображения молекул) или с заготовками моделей молекул. Например, динамицин А и кристалл NaCl (рисунок 2.1).

Рисунок 2.1
· Пункты Open/Close Window/Save/Save As. (Открыть / Закрыть окно / Сохранить / Сохранить как…) представляют собой стандартные функции открытия, закрытия и сохранения файлов.
· Пункт Revert to Saved (Вернуться к сохраненному) убирает все изменения после последнего сохранения.
· Пункт Set Default Settings (По умолчанию) использует установки данной модели для новой модели.
· Пункты Print/Print Setup (Печать / Настройка) открывает стандартное диалоговое окно для настройки и последующей распечатки файла.
Пункт меню Edit (Правка) также предоставляет обычный набор функций копирования, вставки, отмены последнего действия и выбора.
· Пункт Undo/Redo (Отменить / Вернуть) отменяет или, наоборот, возвращает изменения последнего действия.
· Пункт Cut (Вырезать) вырезает выбранный элемент молекулы / текст и копирует его в буфер обмена.
· Пункт Copy (Копировать) копирует выбранный элемент молекулы / текст в буфер обмена.
· Пункт Paste (Вставить) выбранный элемент молекулы / текст из буфера обмена.
· Пункт Clear (Очистить) удаляет выбранный элемент / текст.
· Пункт Copy As (Копировать как…) позволяет копировать выделенное как ChemDraw - структуру или растр (Bitmap)
· Пункт Select All (Выбрать все) выбирает все элементы.
· Пункт Select Atoms (Выбрать атомы) позволяет выбирать атомы по определенному признаку: Revers (Выбрать невыбранные, т.е. обратить выделение), Select C (Выбрать все атомы одного элемента), Select Adjiacent (Выбрать все атомы, смежные к данному), Select Fragment (Выбрать все атомы данного фрагмента).
· Пункты Clear Frames/ Molecular Surfaces/Calculations (Очистить) позволяет очистить кадры / молекулярные поверхности / расчеты.
Пункт меню View (Вид) предоставляет широкий ассортимент настроек вида молекул / связей / панелей и т.д. Он состоит из трех разделов.
Первый раздел включает три пункта: галочку Toolbar (Главная панель), включающую отображение главной панели; галочку Tools Palette (Контрольная панель), отображающую видимость контрольной панели, и пункт Settings (Настройки), выводящий диалоговое окно настройки вида молекул. Его назначение мы рассмотрим позднее.
Второй раздел объединяет пункты, с помощью которых можно визуализировать различные расчетные характеристики молекулярной модели. Подробно Соответствующие расчетные методы компьютерной химии будут рассмотрены в последующих главах. Здесь же мы лишь опишем назначение отдельных пунктов.
• Пункт Solvent Accessible Surface (Поверхность, доступная растворителю) отображает модель молекулы в виде поверхности, доступной молекулам растворителя. Поверхность изображается на основании расчета методом Конолли площади, заметаемой центром пробной сферы (моделирующей растворитерль) при ее мысленном прокатывании по поверхности исследуемой молекулы. При этом атомы в модели молекулы представляются в виде сфер с радиусами, равными ван-дер-ваальсовскимрадиусам соответствующих атомов.
Это окно включает следующий набор настроек:
• Solvent Radius (Радиус растворителя) может быть установлен значениями от 0,1 до 10 А движением бегунка. По умолчанию радиус растворителя установлен равным 1.4 А, соответствующим воде.
• Map Properti (Свойства поверхности) предлагает настройки цвета отображения поверхности.
• Resolution (Разрешение) устанавливает качество отображаемой поверхности от грубого до тонкого. Как видно, при низком разрешении поверхность, доступная растворителю, становиться почти эллипсоидальной, тогда как при высоком разрешении она почти повторяет исходную модель молекулы.
Рисунок 2.2 Модель молекулы бутана (а) и поверхность доступная растворителю с разрешением 40 (б)
· Пункт Connolly Molecular Surface (Молекулярная поверхность Конолли) предлагает аналогичные настройки.Вданном случае для построения поверхности используется другой алгоритм Конолли, в котором поверхность определяется контактом с молекулой растворителя − сферой с заданной величиной ван-дер-ваальсовского радиуса.
· Пункт Total Charge Density (Общая плотность заряда) позволяет визуализировать пространственную плотность распределения заряда − электронную плотность в пространстве, окружающем ядро молекулы, или вероятность нахождения электронов в окружении молекулы.
Окно настроек позволяет задать метод расчета (Calculation), тип поверхности (Surface Туре), ее цвет и т.д.
· Пункт Total Spin Density (Спиновая плотность) описывает пространственное распределение разницы в плотностях между ↑-спиновыми и ↓-спиновыми электронами в данной точке.
· Пункт Molecular Electrostatic Potential (Молекулярный электростатический потенциал) позволяет оценить распределение молекулярного электростатического потенциала. Последний характеризует притяжение или отталкивание между молекулой и протоном в данной точке. Притяжение определяется отрицательными значениями потенциала, а отталкивание - положительными.
· Пункт Molecular Orbitalis (Молекулярные орбитали) позволяет показать поверхности, визуально отображающие различные стабильные электронные распределения в молекуле. Расчет их может быть проведен квантово-химическими методами.
· Пункт Partial Charges (Частичные заряды) используется для отображения частичных зарядов на атомах, вычисленных на основании квантово-химического расчета.
В последнем разделе пункта меню View приведены ссылки па таблицы с параметрами, используемыми в расчетах. Например, таблица ММ2 Atom Type Parameters содержит поля с параметрами атомов, используемыми при расчете энергии молекулы методом молекулярной механики.
Пункт меню Tools (Инструменты) предлагает инструменты отображения различных панелей, меню, функции масштабирования и отражения молекулы.
В первом разделе сосредоточены пункты настройки рабочего окна программы.
· Галочка Show Status Bar включает отображение строки состояния.
· Галочка Show Rotation Bars включает отображение средств вращения молекулы в окне визуализации.
· Галочка Show Movie Controller включает отображение контроллера анимации.
· Галочка Show Messages включает отображения поля с сообщениями.
· Галочки Show Model Table включает отображения полей с различными данными (измерения, группы, координаты)
· Галочка Show H's and Lp's включает отображение атомов водорода и неподеленных электронных пар в модели молекулы.
Рисунок 2.3 Модель молекулы этилового спирта: а) отображение атомов водорода и неподелённых электронных пар; б) выключено
В следующих разделах сосредоточены пункты, позволяющие управлять отображением модели молекулы в окне редактора модели.
• Функции Magnify (Увеличить) и Reduce (Уменьшить) соответственно изменяют масштаб молекулы в окне визуализации. Их действие дублируют кнопки в нижней левой части окна модели, а также горячие клавиши F7 и F8 соответственно.
• Функция Rectify (Исправить) проверяет наличие в молекуле связей, согласно химическим правилам. Например, если четырехвалентный углерод содержит только две связи, будут добавлены два атома водорода. По умолчанию это делается автоматически.
• Функция Clean Up Structure (Очистить) проверяет соответствие в модели молекулы длин связей и валентных углов принятым в химии стандартным значениям. Если соответствие отсутствует (например, когда молекула рисовалась вручную), то делаются необходимые исправления в ее структуре.
• Функция Dock (Стыковать) ориентирует один фрагмент относительно другого.
• Функция Overlay (Покрыть) покрывает один фрагмент другим с помощью атомных пар.
• Пункт Reflect (Отразить) позволяет отразить молекулу относительно заданных плоскостей или инвертировать ее относительно начала координат.
• Пункт Fit Model to Window (Вписать модели в окно) изменяет масштаб молекулы таким образом, чтобы она была полностью видна в окне визуализации.
• Пункт Fit Selection to Window (Вписать выделение в окно) изменяет масштаб выделенной части модели (Selection) таким образом, чтобы она была полностью видна в окне визуализации.
• Пункт Fit All Frames to Window (Вписать все кадры в окно) изменяет масштаб всех кадров анимации таким образом, чтобы они были полностью видны в окне визуализации.
Пункт меню Object (Объект) предлагает инструменты настройки отображения различных параметров молекулы, а также измерения длин связей, межатомных расстояний и углов.
• Функция Move to Center (Двигать в центр) перемещает молекулу или выделение в центр окна визуализации.
• Функция Move То (Двигать в…) перемещает молекулу или выделение на соответствующую ось X, Y или Z.
• Пункт Colorize (Изменить цвет) дает возможность изменить цвет атомов по желанию с помощью палитры.
• Пункт Show Elements Symbols (Показать символы элементов) позволяет отображать символы элементов (например, С, Н и т.д.).
• Пункт Show Serial Numbers (Показать порядковые номера) отображает порядковые номера атомов в модели молекулы, установленные программой.
• Пункт Show Solid Spheres (Показать твердые сферы) включает отображение атомов в виде сплошных сфер.
• Пункт Show Dot Surfaces (Показать точечные поверхности) включает отображение атомов в виде сфер с ван-дер-ваальсовскими радиусами посредством точек.
• Функции Set Distance (Определить расстояние), Set Bond Length (Определить длину связи), Set Bond Angle (Определить угол) и Set Dihedral Angle (Определить двугранный угол) позволяют измерить и при необходимости изменить соответствующие расстояния и углы в модели молекулы. Для того чтобы пользоваться ими, необходимо выделить с помощью мыши атомы, образующие нужную связь или формирующие нужный угол, и выбрать соответствующую функцию. Результат измерения будет отображен в окне Measurements (Измерения) в поле Actual (Фактическое). Кроме того, в поле Optimal будет отображено значение данной величины, принятое в качестве стандартного для выбранной связи или угла. При необходимости можно измерить выделенное расстояние между атомами или угол, следует набрать с клавиатуры требуемое значение в поле Actual и нажать клавишу ввода или щёлкнуть мышью в окне редактора.
• Функция Break Bond (Разорвать связь) разрывает выделенную связь.
• Пункт Set Bond Order (Установить порядок связи) изменяет порядок связи на одинарный / двойной / тройной.
• Функция Bond Proximate (Ближайшая связь) создает химическую связь между двумя ближайшими атомами. Ближайшими в программе считаются два атома, расстояние между которыми меньше, чем принятое стандартное значение для длины связи плюс некоторое значение, величину которого (в процентах от длины связи) можно изменить в настройках пункта меню View\Settings\Building.
• Пункт Add Centroid (Добавить центровой) создает атом-пустышку в центре выделенной молекулы. Эта функция удобна при моделировании координационных соединений.
• Функция Invert (Инвертировать) выполняет операцию пространственной симметрии в выбранной группе хиральных атомов.
• Пункт Set Z Matrix (Установить Z-матрицу) позиционирует выбранный атом в Z-матрице молекулы. Относительное положение каждого атома в молекуле определяется набором переменных, образующих так называемую Z-матрицу молекулы. С помощью данного пункта можно выбрать атом, который в системе отсчета Z-матрицы будет установлен в качестве начального.
• Функции Hide/Show (Скрыть / Отобразить) позволяет скрыть (видимость) выбранных атомов или отобразить их в окне визуализации.
• Пункт Define Group (Определить группу) позволяет сгруппировать выбранные атомы под определенным именем.
Пункт меню Analyze (Анализировать) предлагает инструменты анализа молекул и расчета их свойств.
• Пункты Spin About X/Y/Z/Selected Axis (Вращать) включают режим вращения молекулы относительно оси X/Y/Z или выбранной оси. С помощью кнопок меню анимации возможно записать анимацию, а затем воспроизвести ее.
• Пункт Spin Torsional Angles (Вращать двугранные углы) вызывает операцию вращения определенных групп атомов относительно двугранного угла.
• Пункт Show Measurements (Показать измерения) открывает окно Measurements, в котором отображаются и валентных и двугранных углов для всех атомов молекулы.
• Пункт Extended Huckel Surfaces (Поверхности расширенного метода Хюккеля) позволяет расчитать молекулярную поверхность расширенным методом Хюккеля. Метод Хюккеля − это один из простейших полуэмпирических квантово-химических методов расчёта. Вычисления с его помощью осуществляются наиболее быстро. Следует заметить, однако, что при его использовании не следует забывать о поговорке: «Вам как сосчитать − быстро или точно?» После проведения расчета выберите какую-нибудь молекулярную поверхность (молекулярную орбиталь) в меню View (Вид) и отобразите её кнопкой Show Surface (Показать поверхность). Аналогичным образом можно отобразить распределение частичных зарядов в молекуле.
• Пункт Deviation from Plane (Отклонение от плоскости) показывает отклонения выбранных атомов от нормальной плоскости.
• Пункт Compute Properties (Расчет свойств) вызывает одноименное окно, позволяющее выводить результаты расчета выбранного свойства или группы свойств молекулы или соответствующего вещества. Данный пункт объединяет различные расчетные методы, представленные в пакете Chem3D
Пункты меню ММ2 (Молекулярная механика), Gamess, Gaussian и МОРАС предлагают методы расчета потенциальной энергии молекул и прочих характеристик эмпирическими, полуэмпирическимии и неэмпирическими квантово-механическими методами.
• Пункт Run ММ2 Job… (Запустить ММ2 задачу) открывает диалоговое окно для выбора файла описания задания ММ2 с расширением *.jdf и *.fdt.
• Пункт Minimize Energy… (Минимизировать энергию) запускает процесс минимизациипотенциальной энергии молекулы.
• Пункт Molecular Dynamics… (Молекулярная динамика) активизирует молекулярно-динамический эксперимент с возможностью задания температуры, числа итераций и других параметров.
• Пункт Compute Properties… (Расчет свойств) открывает окно для выбора типа расчета.
• Пункт Show Used Parameters (Показать использовавшиеся параметры) отображает параметры ММ2 в окне Messages (Сообщения).
Контрольная панель
Контрольная панель располагается в левой части экрана и имеет небольшой набор необходимых средств.
• Кнопка (Select Tool) позволяет выбрать индивидуальные атомы или связи. С помощью нажатой кнопки <Shift> можно выбрать группы атомов или связей.
• Кнопка (Trackball Tool) вращает целую модель вокруг осей X и Y.
• Кнопки (Bond) создают одинарную / двойную / тройную химическую связь.
• Кнопка (Uncoordinated bond) создает некоординированную связь (с неспаренным электроном).
• Кнопка (Text) позволяет ввести новый элемент, тип атома, формальный заряд или порядковый номер для выбранного атома молекулы, кроме того, дает возможность заменить тип выбранных атомов. Если щелкнуть по пустому месту в окне визуализации, то будет дана возможность создать новый фрагмент, просто записав формулу, например С2Н50Н.
• Кнопка (Eraser Tool) - средство, позволяющее «стирать» атомы и связи.
Окно визуализации располагается в центре экрана и служит средством создания, редактирования и отображения молекулярных структур.
По краям окна располагаются кнопки вращения молекулы.
• Кнопка (Revolve) вращает всю молекулу целиком или на определённый выбранный угол.
• Кнопки (Scale) позволяют уменьшить / увеличить масштаб изображения в окне визуализации.
• Кнопки (Animation) используются при создании и воспроизведении анимации (рисунок 2.4).
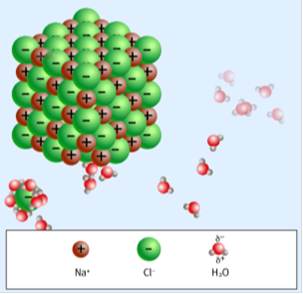
Рисунок 2.4 Фрагмент анимационной компьютерной модели «Диссоциация хлорида»
• Окно Messages (Сообщения), открываемое кнопкой-треугольником, отображает в текстовом виде результаты измерений и вычислений.
Заключение
Последние десятилетия двадцатого века ознаменовались глобальными переменами, затронувшими практически все сферы жизни человечества. Мир вступил в эпоху информационного общества, для которого характерно, прежде всего, перепроизводство научной информации. Наращивание информации в обществе продолжается на беспрецедентном уровне. Многие считают, что в течение ближайших десяти лет 100% того, что мы сегодня знаем, составит лишь 10% имеющийся базы знаний. Постоянно обновляющиеся научные знания требуют от человека, прежде всего, умения ориентироваться в информационном пространстве, критически подходить к отбору информации, создавать новые знания и способы деятельности на основе полученной информации. Кроме того, применение информационных технологий даёт возможность сделать процессы обучения и управления процессами обучения более эффективными и интенсивными.
Подводя черту, под всем выше сказанным хочется заметить, что компьютерные программы, в том числе и ChemOffice, могут успешно применяться как для научной деятельности, так и при изучении курса химии в школе и ВУЗах.
Библиографический список
1. Белохвостов, А.А. К вопросу об использовании информационных технологий в обучении / А.А. Белохвостов // Материалы XVII Международной электронной научной конференции «Новые технологии в образовании». - Воронеж, 2006. - С. 11-12
2. Белохвостов, А.А. Визуализация пространственной структуры химических соединений / А.А. Белохвостов // Материалы региональной научной конференции студентов, магистрантов, аспирантов и молодых учёных. II Машеровские чтения. - Витебск: изд-во «ВГУ им. П.М. Машерова», 2007. - С. 48-49
. Соловьёв, М.Е. Компьютерная химия/ Соловьёв М.Е., Соловьёв М.М. - М.: СОЛОН-Пресс, 2005. - 5 с.
Введение
Создание и совершенствование компьютерной техники, появление новых технологий передачи, обработки, накопления и представления информации облегчило и ускорило вычислительную работу во многих областях науки и, в частности, химии. Квантовая химия, молекулярная механика, планирование химического синтеза, получение и обработка экспериментальных данных с помощью новых информационных технологий и компьютерной техники - это лишь некоторые типичные примеры. Увеличение вклада информационных в развитие химии связано в первую очередь с появлением персональных компьютеров с высокими эксплуатационными характеристиками и новых технологий передачи информации. Благодаря их относительно низкой цене и доступности для широкого круга пользователей такое оборудование стало обычным в большинстве химических лабораторий и учебных заведениях мира.
Сферами использования компьютеров среди химиков является подготовка текстов, содержащих химические структуры, визуализация пространственной структуры молекул, прогнозирование физико-химических свойств органических соединений на основании их химического строения и т.д. Популярные текстовые редакторы, такие как Microsoft Word, Word Perfect или WordPro и др., несмотря на свои широкие возможности, не имеют удобных инструментов для создания химических структур высокого качества. Для решения задач создания химических структур, схем реакций в полиграфических целях, использования их при создании баз данных, поиска в информационных системах, программах квантовой химии и других областях создано много различных программных средств. Одной из них является - ChemOffice.
Интегрированный программный комплекс ChemOffice включает следующие четыре специализированных приложения:
1) «химический редактор» CS Сhem Draw, являющийся традиционным средством редактирования химических формул;
2) специальный редактор баз данных СS СhemFinder, предназначенный для создания, редактирования и управления базами данных химических соединений;
) программа CS Chem3D, предназначенная для визуализации химических соединений, компьютерного моделирования и расчётов;
) редактор таблиц CS Table Editor, предназначенный для просмотра и редактирования табличных данных, используемых в пакете CS Chem3D.
Одним из важнейших элементов химического исследования является анализ геометрической структуры соединений. Эта область науки получила название структурная химия. Важнейшими экспериментальными методами, позволяющими исследовать геометрическую структуру молекул, является адсорбционная и эмиссионная спектроскопия. А также дифракционные методы. Структурные формулы отражают связанность различных атомов и молекул друг с другом. Классическим примером двухмерной структурной модели является структурная формула бензола: Брутто-формула С6Н6 не позволяет передать взаимосвязь между атомами углерода кольца, в связи с чем возникает необходимость отображения структуры в виде геометрического образца - задача визуализации структуры. Ещё более актуальной эта задача становиться при изучении стереоизомеров. Конформационный анализ соединений требует использование трёхмерной модели молекул. До появления компьютеров в качестве таких моделей широко применялись механические модели молекул (модели Стьюарта-Бриглеба). За последние десятилетие создано большое количество различных программных пакетов, позволяющих решать задачи визуализации как плоских, так и пространственных моделей молекул. Первые версии программ позволяли решать в основном задачи пространственных структур. В дальнейшим круг задач, решаемых с использованием специализированных «химических» программных пакетов, был расширен, и в настоящее время наиболее мощные из них реализуют практически все основные задачи компьютерной химии, включая управление базами данных химических соединений, методы квантовой химии и численного моделирования.
Целью данной работы было изучить возможности программного комплекса СhemOffice для построения, корректирования и двухмерной и трёхмерной визуализации химических формул.
В связи с поставленной целью нами решались следующие задачи:
1) Описать программное приложение ChemDraw 8.0 и выявить его возможности для двумерного построения и редактирования структурных химических формул;
2) Описать программное приложение Сhem3D Ultra 8.0 и определить его возможности для трёхмерной визуализации химических формул;
3) Определить различные способы редактирования и анализа геометрии трехмерных моделей молекул.
Редактирование структурных химических формул в программе ChemDraw

Типы оградительных сооружений в морском порту: По расположению оградительных сооружений в плане различают волноломы, обе оконечности...

Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...

Археология об основании Рима: Новые раскопки проясняют и такой острый дискуссионный вопрос, как дата самого возникновения Рима...

Папиллярные узоры пальцев рук - маркер спортивных способностей: дерматоглифические признаки формируются на 3-5 месяце беременности, не изменяются в течение жизни...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!