Электрофильные реакций в основном органическом и нефтехимическом синтезе большое значение имеют процессы присоединения по ненасыщенным углерод-углеродным связям и замещения при атоме углерода в ароматическом ядре. Из первых можно отметить аддитивное хлорирование и гидратацию олефинов и ацетилена, присоединение кислот и других веществ к ним, ионную полимеризацию олефинов, алкилирование изопарафинов. Важнейшими из реакций ароматических соединений являются их хлорирование, нитрование, сульфирование, алкилирование. По сравнению с относительно мало распространенными нуклеофильными реакциями ненасыщенных и ароматических соединений электрофильные превращения этих веществ типичны в процессах их промышленной переработки.
12.1. Электрофильное присоединение по С = С‒связи
За счет π-электронов на двойной связи в молекулах олефинах сосредоточена повышенная электронная плотность. Поэтому связь С=С склонна подвергаться атаке электрофильным (электронодефицитным) реагентом. В этом случае будет происходить гетеролитический разрыв π-связи, и реакция пойдет по механизму электрофильного присоединения, который обозначается символом AdE (от англ. Addition Electrophilic).
Реакция электрофильного присоединения по ненасыщенным углерод-углеродным связям протекает по схеме

На первой стадии электрофил присоединяется по двойной связи олефина с образованием карбкатиона. На второй стадии карбкатион взаимодействует затем с каким-либо нуклеофилом и дает целевой продукт.
Стадия присоединения электрофила по двойной связи не является одностадийной ‒ присоединению а предшествует образование π-комплекса электрофила с олефином. Олефины, как и другие ненасыщенные соединения, могут связываться с электрофилом за счет частичного размещения пары π-электронов кратной связи на свободной орбитали электрофила (рис. 12.1)

Рис. 12.1. Схема образования π-комплекса электрофила с олефином
Акцепторами электронов π‒связи кроме 1 s ‒орбитали протона могут бытьсвободные орбитали в молекулах кислот Льюиса, например, 2 р ‒орбиталь азота в NO2+ или ионах карбония,4 р ‒орбиталь в молекуле Вr2 и т. д.:

Все эти соединения, получившие название π‒комплексов, образуются по быстрым равновесным реакциям и легко распадаются на исходные компоненты. Дальнейшее превращение π‒комплекса в карбкатион происходит по медленной мономолекулярной реакции, в результате чего электроны π‒связи полностью переходят на свободные орбитали одного из атомов углерода и электрофила и образуется σ‒связь С–Е или С–Н.
На рис. 12.2 показано, как изменяется энергия на различных стадиях реакции.

Рис. 12.1. Изменения энергии на различных стадиях электрофильного присоединения по С=С-связи
При взаимодействии иона лиония SH2+ с олефином карбкатион образуется по схеме:

В ряде случаев при образовании π‒комплекса полной передачи протона не происходит, и реакция с кислотой НА протекает по следующей схеме:

Так как ни первая стадия образования π‒комплекса, ни последующее взаимодействие карбкатиона с нуклеофилом не являются лимитирующими, то общая скорость процессов электрофильного присоединения описывается кинетическим уравнением второго порядка

Экспериментально определяемая константа скорости k = К 1 · k 2. возрастает с увеличением кислотности иона лиония SH2+ или молекулы НА. В первом случае это вызвано увеличением константы равновесия К 1, а во втором ‒ увеличением констант К 1 и k 2.
Так, скорость присоединения галогеноводородов к олефинам в апротонных растворителях возрастает в ряду HF < HC1 < НВr < <НI, соответствующему увеличению их кислотности. Эти реакции ускоряются кислотами Льюиса (А1С13, SnCI4, TiCI4). Механизм катализа заключается или в увеличении кислотности галогеноводородов в результате связывания аниона галогена в комплекс с кислотой Льюиса, например

или в воздействии кислоты Льюиса на первоначально образовавшийся π‒комплекс, что ускоряет переход π‒комплекса в ион карбония

В обоих случаях скорость реакции описывается кинетическим уравнением третьего порядка:

В отличие от кислот с достаточно высокой кислотностью присоединение слабых электрофилов протекает только в присутствии каталитических добавок сильных протонных кислот. Так, для присоединения воды к олефинам в качестве катализаторов используют серную или фосфорную кислоту

По такому же механизму к олефинам присоединяются сероводород, меркаптаны, спирты, фенолы, карбоновые кислоты и т. п. Скорость всех этих реакций определяется скоростью образования карбкатиона и пропорциональна кислотности среды и концентрации олефина.
В отсутствие сильных нуклеофилов их роль может выполнять сам олефин, реагируя с карбкатионом. В результате происходит кислотная полимеризация олефинов, идущая с образованием низших полимеров (димеров, тримеров и т. д.):

Полимеризация олефинов уже включает стадию присоединения по двойной связи не только протона, но и карбкатиона, и является промежуточной ко второй группе механизмов катализа реакций электрофильного присоединения, для которых характерно предварительное образование катиона из второго реагента с его последующим присоединением по двойной связи. Примером может служить взаимодействие формальдегида с изобутиленом по реакции Принса, когда карбкатион получается из альдегида при кислотном катализе процесса:

При галогенировании олефинов молекулы галогенов сами способны к образованию π‒комплексов, которые по медленной мономолекулярной реакции превращаются в карбкатионы:

Карбкатион быстро реагирует далее с анионом, образуя целевой продукт галогенирования:

Лимитирующей во всех этих реакциях с галогенами также является стадия перехода π‒комплекса в ион карбония. Скорости реакции описывается в этом случае кинетическими уравнениями второго порядка:

В полярной среде скорость реакции увеличивается благодаря сольватации π‒комплекс. В малополярных средах отщеплению галоген-аниона благоприятствуют кислоты Льюиса, каталитическое действие которых проявляется двояко, в том числе и путем предварительного образования комплекса с галогеном:


Скорость реакции описывается в этом случае кинетическим уравнением третьего порядка:

При реакции с карбкатионами следовало бы ожидать образование равных количеств стереоизомеров. Однако в ряде случаев обнаруживается транс-присоединение к олефинам. Эти факты можно объяснить существованием и таких механизмов реакции, в которых лимитирующей стадией является бимолекулярное взаимодействие π‒комплекса с нуклеофилом:

Реакции транс‒присоединения, особенно часто встречаемые при галогенировании, объясняют также образованием «неклассических» ионов карбония, в которых сохраняется жесткая трехцентровая связь, препятствующая свободному вращению вокруг С–С‒связи:

Имеются также стереоспецифические реакции цис‒присоединения, например, эпоксидирование олефинов надкислотами:

12.2. Электрофильное присоединение при катализе солями переходных металлов и их комплексами
Особую группу составляют реакции, катализируемые комплексами переходных металлов. В большинстве этих реакций первичной стадией является образование π‒комплекса олефина с переходным металлом, которое протекает 1) путем прямого присоединения металла или 2) путем вытеснения из координационной сферы комплекса другого лиганда:

В последнем случае образование π‒комплекса происходит сравнительно медленно, и скорость этого процесса может оказаться сравнимой со скоростью других стадий. Это обусловливает значительную сложность кинетики таких реакций.
Связь органической молекулы с атомом металла осуществляется за счет взаимодействия π‒электронов ненасыщенного соединения с вакантной р ‒орбиталью центрального атома металла. Строение такого комплекса имеет ряд характерных особенностей, обусловленных структурой электронной оболочки центрального атома и природой связанных с ним молекул и атомов лигандов (X). Кроме вакантных р ‒орбиталей переходные металлы имеют частично или полностью заполненные d ‒орбитали. Электроны d ‒орбиталей способны взаимодействовать с вакантными разрыхляющими π‒орбиталями ненасыщенных соединений с образованием, так называемой дативной связи.
Образование как донорно-акцепторной, так и дативной связи приводит к разрыхлению и к уменьшению кратности связи в молекуле ненасыщенного соединения и способствует последующей реакции присоединения. Возникающие при координации полярные эффекты при донорно-акцепторном и дативном взаимодействии противоположны. Донорно-акцепторное взаимодействие приводит к смещению электронов к атому металла и к появлению эффективных положительных зарядов на атомах углерода. При дативном взаимодействии пребывание d-электронов металла на разрыхляющей орбитале ненасыщенного соединения приводит к обратному эффекту.
Способность молекулы ненасыщенного соединения к последующему взаимодействию с тем или иным реагентом во многом определяется соотношением между вкладами дативного и донорно-акцепторного взаимодействия. В настоящее время, однако, отсутствуют надежные критерии для их количественной оценки. Можно лишь отметить, что дативная способность одного и того же элемента в общем возрастает с уменьшением степени его окисления и с увеличением числа отрицательно заряженных лигандов, связанных с металлом. В противоположном направлении изменяются при этом донорно-акцепторные свойства металла. Изменяя природу и число лигандов, связанных с металлом, можно направленно влиять на взаимодействие последнего с ненасыщенным соединением, которое определяет, в конечном счете, скорость и селективность последующих превращений π‒комплекса.
Например, при гидратации ацетилена по Кучерову первичный π‒ комплекс с катионом ртути подвергается внешней атаке по активированному углеродному атому ацетилена молекулой воды с одновременным превращением в нестабильное металлоорганическое σ‒комплексное соединение. Далее проходит протолиз связи С‒Н иизомеризация в ацетальдегид.

С увеличением числа отрицательных лигандов, связанных с центральным атомом ртути в ряду HgCl+ > HgCl > HgCl3, каталитическая активность сильно уменьшается. Это связано с усилением дативного взаимодействия при образовании π‒комплекса с ацетиленом в этом же ряду, что уменьшает эффективный положительный заряд на углероде и препятствует нуклеофильному присоединению воды.
Введение некоторых лигандов может сильно влиять и на направление реакции. Так, реакция присоединения молекулы HCN к ацетилену, проводимая в концентрированном водном растворе солей Cu2Cl2 + NH4Cl, протекает по схеме

Образование π‒комплекс, при добавке некоторых меркаптанов почти полностью подавляется, и ацетилен начинает присоединять только воду с образованием ацетальдегида. Этот эффект объясняют образованием комплекса меди с меркаптаном, который имеет совершенно иные каталитические свойства. Подобные эффекты резкого изменения селективности связаны, по-видимому, кроме электронных, и со стерическим влиянием вводимых лигандов.
12.3. Реакционная способность ненасыщенных веществ, правила присоединения и побочные реакции
При реакциях электрофильного присоединения, идущих спромежуточным образованием карбкатионов, константа скорости, как мы видели выше, равна произведению константы равновесия при образовании π‒комплекса на константу скорости превращения π‒комплекса в карбкатион. При всех изменениях в строении ненасыщенного вещества, приводимых к увеличению этих констант, реакции электрофильного присоединения будут ускоряться. Равновесие образования π‒комплекса зависит от электронной плотности π‒связи: повышение ее облегчает смещение соответствующих электронов в направлении свободной орбитали электрофила. Стадия перехода π‒комплекса в ион карбония, лимитирующая скорость многих реакций электрофильного присоединения. Все электронные эффекты, стабилизирующие этот карбкатион, будут ускорять его образование, что приводит к такому же влиянию заместителей, как и при образовании π‒комплексов.
Следовательно, электронодонорные заместители должны ускорять реакцию электрофильного присоединения, а электроноакцепторные — замедлять ее. Этот эффект противоположен тому, который наблюдается при нуклеофильном присоединении к ненасыщенным веществам. Он может служить одним из доказательств электрофильного механизма реакции. В соответствии с этим, гомологи этилена реагируют быстрее самого этилена, причем наблюдается следующий ряд реакционных способностей:

Так, гидратация изобутилена происходит в 8000 раз быстрее, чем гидратация пропилена, а последний гидратируется в 8000 раз быстрее этилена. Положительно влияет на скорость реакции фенильная группа благодаря сопряжению с реакционным центром, причем стирол гидратируется в 5000 раз быстрее этилена.
Ненасыщенные вещества с электроноакцепторными группами располагаются по реакционной способности в следующий ряд

причем многие из перечисленных ранее реакций электрофильного присоединения с такими соединениями вообще не идут.
При реакциях присоединения, в которых классический ион карбония не образуется (например, при галогенировании), положительный заряд оказывается рассредоточенным между несколькими атомами. На стабилизацию такого карбкатиона заместители имеют уже меньшее влияние, поэтому реакционная способность ненасыщенных веществ различается в этом случае не так сильно. Это можно проиллюстрировать данными по бромированию олефинов

Электронными эффектами заместителей определяется и правило электрофильного присоединения к несимметричным олефинам. С формальной точки зрения, возможны два направления присоединения, например:

Согласно правилу Марковникова, присоединение молекулы НХ идет таким образом, что водород связывается с наиболее гидрированным атомом углерода (направление I). Это правило применимо для всех реакций электрофильного присоединения НХ к алкилзамещенным олефинам. Рассмотрим причины такой закономерности.
Стабильность карбкатиона, как известно, повышается при наличии алкильных заместителей у его положительно заряженного углеродного атома и быстро возрастает в ряду: первичный < вторичный < третичный. По этой причине продукт I получается с 99%-ым выходом. При электрофильных превращениях гомологов этилена из углеводородов с прямой цепью всегда образуются вторичные производные, а из разветвленных — третичные;

На основании изложенного, правило Марковникова можно сформулировать следующим образом: к несимметричным олефинам электрофилы присоединяются с промежуточным образованием более стабильного карбкатиона. При такой формулировке область применимости правила расширяется, оно распространяется на все электрофилы и ненасыщенные вещества с любыми заместителями.
При наличии электроноакцепторных заместителей вторичный карбкатион не обязательно оказывается более стабильным. Такие группы, как ‒CF3, (СН3)3N+ и ‒СООН, благодаря сильным электроноакцепторным свойствам делают образование положительного заряда на соседнем с ними углеродном атоме энергетически невыгодным, и реакции идут против правила Марковникова в его первоначальной формулировке:

Галогены как электроотрицательные заместители занимают промежуточное положение: дезактивируя двойную связь, они в то же время способствуют присоединению по правилу Марковникова, так как α-ион карбония оказывается более стабильным из-за влияния метильной группы и сопряжения с галогеном:

Изложенные правила присоединения и порядок изменения реакционной способности олефиновых соединений в общем сохраняются и для ацетиленов, однако эти закономерности могут существенно изменяться при катализе реакции солями и комплексами переходных металлов. В этом случае из-за существенной роли дативного взаимодействия при активировании молекулы олефина или ацетилена электронодонорные заместители могут замедлять реакцию. В том же направлении действуют также стерические затруднения при вхождении молекулы олефина с объемистыми заместителями в координационную сферу центрального атома переходного металла. В результате для некоторых реакций наблюдается аномальный порядок изменения реакционной способности.
12.4. Побочные реакции и состав продуктов
Кроме специфических для каждой из реакций путей образования побочных продуктов имеются две общие причины их появления: 1) изомеризация промежуточных ионов карбония и 2) наличие в реакционной массе нескольких нуклеофилов, способных взаимодействовать с ионами карбония или соответственно с π‒ и σ‒комплексами координационных соединений переходных металлов.
Первая из этих реакций уже рассматривалась для процессов нуклеофильного типа, идущих через образование карбкатионов, которое еще более характерно для электрофильного присоединения. В результате изомеризации ионов карбония часто получаются смеси продуктов или вещество, строение которых не соответствует исходным реагентам. При этом изомеризация всегда идет в направлении более стабильных ионов карбония с преимущественным образованием соответствующих им продуктов реакции. Типичным примером является алкилирование изопарафинов олефинами.
В случае изобутана и н -бутиленов вначале могут образоваться следующие ионы карбония:

В результате быстрой перегруппировки оба иона переходят в более стабильный третичный карбкатион и при отрыве гидрид‒иона от молекулы изобутана дают главным образом 2,2,4-триметил-пентан:
 Для многих реакций изомеризация ограничивается изменением положения реакционного центра с сохранением углеродного скелета олефина. Например, образование смеси вторичных замещенных при реакциях электрофильного присоединения к н-олефинам:
Для многих реакций изомеризация ограничивается изменением положения реакционного центра с сохранением углеродного скелета олефина. Например, образование смеси вторичных замещенных при реакциях электрофильного присоединения к н-олефинам:

При реакциях галогенирования, идущихс промежуточным образованием «неклассических» ионов карбония, такой изомеризации обычно не происходит.
12.5. Реакции олефинов
Оксосинтез альдегидов из олефинов, окиси углерода и водорода протекает при давлении 10‒30 МПа при катализе кобальтом или его солями, например, дикобальтоктакарбонил Со2(СО)8.
Дикобальтоктакарбонил Со2(СО)8 дает с водородом гидрокарбонил кобальта НСо(СО)4, которому отводится основная роль в катализе реакций оксосинтеза. Гидрокарбонил, вытесняя одну молекулу СО, образует π‒ комплекс с олефином, который затем превращается в металлоорганический σ‒комплекс:

Из-за обратимости этих реакций при высоком парциальном давлении окиси углерода концентрация π‒ и σ‒комплексов снижается, что замедляет процесс в таких условиях. Дальнейшее-взаимодействие внутри комплекса также включает стадию внедрения молекулы СО между связью Со—С:
 Последующее разрушение С‒Co‒связи водородом или гидрокарбонилом кобальта ведет в итоге к образованию альдегида:
Последующее разрушение С‒Co‒связи водородом или гидрокарбонилом кобальта ведет в итоге к образованию альдегида:

Интересен механизм практически важной реакции окисления олефинов в присутствии хлористого палладия. Эти вещества взаимодействуют по следующей реакции:

Если в каталитической системе присутствует и соль двухвалентной меди, то медь окисляет палладий, асама при пропускании воздуха или кислорода непрерывно регенерируется:

Сложение всех уравнений дает суммарную реакцию:

Изучение механизма реакции, осуществляемой в слабокислой среде, показало, что процесс идет через координационные соединения – π‒ и σ‒комплексы палладия, присутствующего в растворе главным образом в виде аниона PdCl42-. Сначала олефин и вода последовательно входят в координационную сферу палладия, заменяя ионы хлора по обратимым реакциям

Затем координационно связанная вода отдает протон растворителю, а образовавшийся сильный нуклеофил НО– атакует углеродный атом олефина содновременным превращением π-комплекса в σ‒комплекс:

Предыдущие обратимые реакции обусловливают замедление процесса при повышении кислотности среды и концентрации анионов хлора. σ‒Комплекс претерпевает внутреннее превращение смиграцией гидрид-иона, разрушающей металлоорганическую связь:

Образовавшийся карбкатион отщепляет протон и переходит в ацетальдегид (в случае других олефинов ‒ в кетон).
12.6. Электрофильное замещение в ароматических соединениях
12.6.1. Механизм реакций электрофильного замещения в ароматических соединениях
Среди многих реакций этого типа в основном органическом и нефтехимическом синтезе главное значение имеют алкилирование, сульфирование, хлорирование и нитрование:
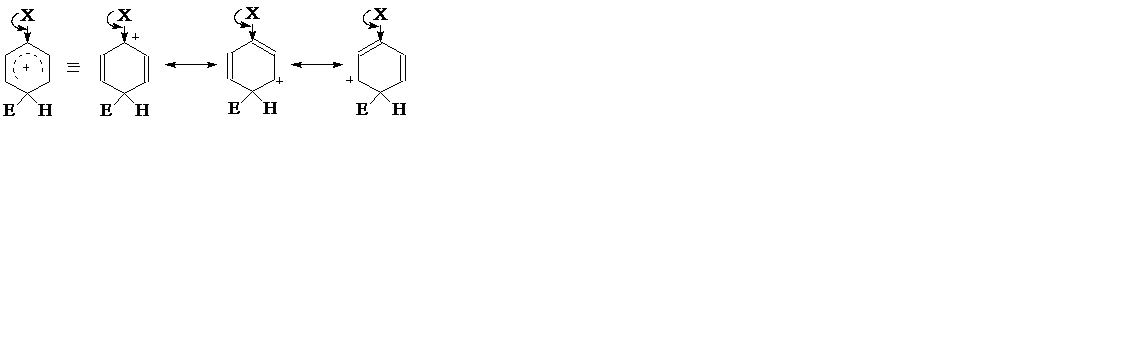
Их механизм имеет общие черты и во многом сходен с механизмом электрофильного присоединения по кратным связям. Реакция начинается с образования π‒, а затем σ‒комплекса ароматического соединения с электрофилом Е+:

Положительный заряд σ‒комплекса не локализован на каком-либо одном углеродном атоме, как в обычном карбкатионе, а распределен по ядру. Несмотря на это, σ‒комплексы являются такими же энергетически нестабильными и высоко реакционноспособными соединениями, как и карбкатионы с локализованным положительным зарядом.
Для дальнейшего взаимодействия σ‒комплекса с основанием В имеются две возможности — присоединение (как в реакциях электрофильного присоединения по кратным связям) или отрыв протона с образованием продукта замещения. В случае ароматических соединений реакция идет только по второму направлению

что обусловлено значительно большей энергетической стабильностью ароматической системы по сравнению с производными дигидробензола.
Рассмотрим кинетику процесса электрофильного замещения. Опуская быструю равновесную стадию образования π‒комплекса, которая не влияет на вид кинетического уравнения, процесс электрофильного замещения можно изобразить следующей схемой:

Применив принцип стационарности к концентрации σ‒комплекса получим:

Это выражение упрощается в двух предельных случаях. Если отрыв протона происходит намного быстрее распада σ‒комплекса на исходные компоненты (k 2[B]>> k -1), величиной k -1в знаменателе можно пренебречь, и скорость электрофильного замещения будет равна скорости образования σ‒комплекса:

Наоборот, при противоположном соотношении скоростей реакций распада σ‒комплекса и отрыва протона (k -1<< k 2[B])кинетическое уравнение примет такой вид:
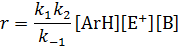
Поскольку в реакциях электрофильного замещения в роли основания В выступает, как правило, находящийся в избытке растворитель, экспериментальное кинетическое уравнение в обоих случаях имеет одинаковый вид

что не позволяет сделать выбор между механизмами реакции.
Ответ на вопрос о лимитирующей стадии процесса можно получить путем измерения кинетического изотопного эффекта. Суть этого эффекта заключается в том, что при замене отщепляемого водорода дейтерием константы скоростей реакций, при которых происходит разрыв связи С–Н или С–D, уменьшаются в 2‒3 раза(k Н/ k D = 2‒3), а при замене тритием Т – в 10‒20 раз (k Н/ k Т = 10‒20). Это связано с большей энергией связей С–D и С–Т по сравнению с энергией связи С–Н. Экспериментально определяемые константы скоростей в предыдущих уравнениях по-разному зависят от константы скорости отрыва протона (k 2 ), и по величине кинетического изотопного эффекта (k Н/ k D или k Н/ k Т)можно сделать выбор между ними. Как показали специально проведенные исследования, величина кинетического изотопного эффекта (k Н/ k Т) для большинства реакций электрофильного замещения не превышает 1,3, т. е. изменение k 2 в 10–20 раз вызывает лишь незначительное изменение экспериментальной константы скорости. Это говорит о том, что k 2[B] > > k –1, и скорость процесса электрофильного замещения практически описывается кинетическим уравнением для реакции образования σ-комплекса. Довольно большой кинетический изотопный эффект был найден только при некоторых реакциях сульфирования. В этих реакциях скорости отрыва протона и распада σ‒комплекса оказались соизмеримы друг с другом (k Н/ k D = 1,6 – 1,8). Однако при увеличении основности и концентрации основания В произведение k 2[В] становится больше k –1 и кинетический изотопный эффект не обнаруживается и в этих реакциях.
В действительности кинетика электрофильного замещения является более сложной, так как на нее оказывает влияние стадия образования электрофила, специфичная для каждой реакции.
12.6.2. Реакционная способность и направление реакций электрофильного замещения в ароматических соединениях
Как установлено выше, скорость реакций электрофильного замещения определяется стадией превращения π‒комплекса в σ‒комплекс. Все электронные и стерические эффекты, стабилизирующие σ‒комплекс, будут способствовать реакции. Этим определяются относительная реакционная способность ароматических соединений и направление замещения в орто ‒, мета ‒и пара ‒положения замещенных бензолов. Ориентирующее влияние заместителей оценивается с помощью факторов парциальных скоростей.
Фактор парциальной скорости – это отношение скорости реакции замещения в орто ‒, мета ‒ и пара ‒положении молекулы С6Н5Х к скорости замещения в одном из шести положений самого бензола. Эти факторы рассчитывают из относительной реакционной способности данного ароматического соединения по сравнению с бензолом (Rt= ki / k 0)и состава получаемых изомеров с учетом того, что в монозамещенном бензоле имеются по два орто‒ и мета‒положения и одно пара ‒положение:
 U Myj/M2jKjTCWtvFvgXN2qmh0mIFKaON+VzXsrlvlYz7Id2vWaJ6bZp9eJgVgpZLC0/rHnb19T/Cb n3T1AwAA//8DAFBLAwQUAAYACAAAACEAdtcb698AAAAKAQAADwAAAGRycy9kb3ducmV2LnhtbEyP TU+DQBCG7yb+h8008WaXEkIssjSNtYkmvVg8eNyyU6BlZwm7pfjvHeOh3ubjyTvP5KvJdmLEwbeO FCzmEQikypmWagWf5fbxCYQPmozuHKGCb/SwKu7vcp0Zd6UPHPehFhxCPtMKmhD6TEpfNWi1n7se iXdHN1gduB1qaQZ95XDbyTiKUml1S3yh0T2+NFid9xerYDwlG+t2x/dtufsq15u3M8XJq1IPs2n9 DCLgFG4w/OqzOhTsdHAXMl50CuIkWjKqIEkXIBj4Gxy4SJcpyCKX/18ofgAAAP//AwBQSwECLQAU AAYACAAAACEAtoM4kv4AAADhAQAAEwAAAAAAAAAAAAAAAAAAAAAAW0NvbnRlbnRfVHlwZXNdLnht bFBLAQItABQABgAIAAAAIQA4/SH/1gAAAJQBAAALAAAAAAAAAAAAAAAAAC8BAABfcmVscy8ucmVs c1BLAQItABQABgAIAAAAIQD+J0pw6wEAAOwDAAAOAAAAAAAAAAAAAAAAAC4CAABkcnMvZTJvRG9j LnhtbFBLAQItABQABgAIAAAAIQB21xvr3wAAAAoBAAAPAAAAAAAAAAAAAAAAAEUEAABkcnMvZG93 bnJldi54bWxQSwUGAAAAAAQABADzAAAAUQUAAAAA " strokecolor="black [3040]" strokeweight="1pt"/> U Myj/M2jKjTCWtvFvgXN2qmh0mIFKaON+VzXsrlvlYz7Id2vWaJ6bZp9eJgVgpZLC0/rHnb19T/Cb n3T1AwAA//8DAFBLAwQUAAYACAAAACEAdtcb698AAAAKAQAADwAAAGRycy9kb3ducmV2LnhtbEyP TU+DQBCG7yb+h8008WaXEkIssjSNtYkmvVg8eNyyU6BlZwm7pfjvHeOh3ubjyTvP5KvJdmLEwbeO FCzmEQikypmWagWf5fbxCYQPmozuHKGCb/SwKu7vcp0Zd6UPHPehFhxCPtMKmhD6TEpfNWi1n7se iXdHN1gduB1qaQZ95XDbyTiKUml1S3yh0T2+NFid9xerYDwlG+t2x/dtufsq15u3M8XJq1IPs2n9 DCLgFG4w/OqzOhTsdHAXMl50CuIkWjKqIEkXIBj4Gxy4SJcpyCKX/18ofgAAAP//AwBQSwECLQAU AAYACAAAACEAtoM4kv4AAADhAQAAEwAAAAAAAAAAAAAAAAAAAAAAW0NvbnRlbnRfVHlwZXNdLnht bFBLAQItABQABgAIAAAAIQA4/SH/1gAAAJQBAAALAAAAAAAAAAAAAAAAAC8BAABfcmVscy8ucmVs c1BLAQItABQABgAIAAAAIQD+J0pw6wEAAOwDAAAOAAAAAAAAAAAAAAAAAC4CAABkcnMvZTJvRG9j LnhtbFBLAQItABQABgAIAAAAIQB21xvr3wAAAAoBAAAPAAAAAAAAAAAAAAAAAEUEAABkcnMvZG93 bnJldi54bWxQSwUGAAAAAAQABADzAAAAUQUAAAAA " strokecolor="black [3040]" strokeweight="1pt"/> 
| 


|
Например, в уксусном ангидриде при 30°С толуол нитруется в 27 раз быстрее бензола, и образуется 58,1 % орто ‒, 38,2% пара ‒ и 3,7% мета ‒ нитротолуола, следовательно факторы парциальных скоростей равны.



Факторы парциальных скоростей дают очень важную информацию о двух тесно связанных между собой аспектах реакционной способности ‒ межмолекулярной (субстратной) и внутримолекулярной (позиционной) селективности.
Межмолекулярная селективность определяет избирательность конкретного электрофила по отношению к различным замещенным бензолам. Некоторые электрофилы обладают высокой межмолекулярной селективностью и для них наблюдается большие различия в скоростях замещения в зависимости от природы заместителя в кольце. Для других реагентов межмолекулярная селективность, напротив, весьма низка. Низкая межмолекулярная селективность характерна для сильных электрофилов, в то время как слабые электрофилы проявляют высокую межмолекулярную селективность.
Внутримолекулярная селективность замещения в орто‒, мета‒ и пара ‒положения, также зависит от природы электрофила. Фактор парциальной скорости отражает связь внутримолекулярной селективностью с природой электрофильного агента. Слабые электрофилы характеризуются и высокой внутримолекулярной селективностью. Реакционноспособные электрофильные агенты проявляют низкую внутримолекулярную селективность.
Для количественного описания позиционной селективности электрофилов предложен еще один параметр – фактор селективности (Sf), представляющий собой логарифм отношения fп / fм

Высокое значение Sf также как и высокое значение fп характерны для слабый электрофилов, проявляющих очень высокую меж‒ и внутримолекулярную селективность. Низкие значения Sf и fп типичны для очень реакционноспособных.
 Рис. 12.2. Зависимость между реакционной способностью (lg f п) и селективностью (Sf) различных реакций замещения в толуоле Рис. 12.2. Зависимость между реакционной способностью (lg f п) и селективностью (Sf) различных реакций замещения в толуоле
| Оказалось, что в случае толуола для разных электрофильных реакций наблюдается линейное отношение между этими величинами, причем тангенс угла наклона прямой лишь немного больше единицы (рис. 12.2). Необходимо отметить, что прямая проходит через начало координат. Эта точка соответствует статистическому распределению орто-, мета и пара -изомеров, которое не достигается ни для одного из известных в настоящее время активных электрофильных агентов. Для других замещенных бензолов также должны получиться аналогичные зависимости, но с другим наклоном и расположением прямых
|
Обычно все заместители X в ароматическом ядре подразделяют на три группы: 1) opтo ‒ пapa ‒ориентанты, активирующие ядро; 2) орто ‒ пара ‒ориентанты, дезактивирующие ядро; 3) мета ‒ориентанты, дезактивирующие ядро. В действительности резкая грань между ними отсутствует, и для каждого замещенного бензола образуются в разных пропорциях все возможные изомеры.
1. Активирующие opтo ‒ пара ‒ориентанты активируют все или по крайней мере орто ‒и пара ‒положения замещенного ароматического соединения по сравнению с бензолом; при этом особенно сильно ускоряется замещение в орто- и пара ‒положениях. Такой эффект проявляют алкильные и арильные заместители (‒СН3, ‒С2Н5, ‒С6Н5), заместители с кислородсодержащими группами (–ОН, –О–, –OR, –ОС6Н5) и аминогруппами (–NH2, –NR2). Например, при бромировании толуола в среде уксусной кислоты факторы парциальной скорости равны: fп = 2420, fм = 5,5, fо = 600. Общее ускорение реакции замещения связано с электронодонорным эффектом перечисленных групп, повышающим стабильность промежуточных σ‒комплексов. При этом алкильные группы оказывают такое влияние за счет положительного индуктивного эффекта и сверхсопряжения с α‒С–Н-связями, а остальные ‒ за счет сопряжения их р ‒или π‒электронов с ароматической системой связей, превышающего отрицательный индуктивный эффект некоторых из этих групп:

При этом больше всего стабилизируются орто- и пара ‒σ‒комплексы, что наиболее наглядно можно объяснить исходя из их резонансных структур, наложение которых дает действительное строение пара ‒σ‒комплекса:
Среди трех резонансных структур первая существенно стабилизирована электронодонорным заместителем, компенсирующим ее положительный заряд. Эта структура приобретает поэтому большую роль в действительном строении σ‒комплекса, обусловливая его повышенную стабильность. Подобная структура имеется и для орто ‒σ‒комплекса

Для мета ‒положения группа X и положительный заряд в резонансных структурах удалены друг от друга:

Здесь стабилизация σ‒комплексов может достигаться только за счет индуктивного эффекта.
2. Дезактивирующие орто‒пара ‒ориентанты уменьшают скорость замещения во всех положениях ароматического ядра по сравнению с бензолом; особенно сильно дезактивируется при этом мета‒ положение.
К заместителям этой группы относятся галогены, а также винильные группы типа ‒CH =CHR. Реакция нитрования хлорбензола характеризуется, например, следующими факторами парциальных скоростей: fо = 0,029, fп = 0,137, fм = 0,0009. Замедление замещения во всех положениях объясняется снижением общей электронной плотности ароматического ядра благодаря электроноакцепторным свойствам этих заместителей. При этом дезактивирование орто‒ и пара ‒положений меньше, чем мета‒, из-за эффекта сопряжения в орто‒ и napa ‒замещенных σ