Нуклеофильной называется реакция, в которой молекула органического вещества подвергается действию нуклеофильного реагента.
Нуклеофильные ("любящие ядро") реагенты или нуклеофилы - это частицы (анионы или молекулы), имеющие неподеленную пару электронов на внешнем электронном уровне. К нуклеофилам относятся НО-, Cl-, CN-, вода, спирты, аммиак и амины.
Нуклеофильные реакции имеют важное значение в промышленности основного органического и нефтехимического синтеза. К их числу относятся процессы замещения и расщепления галогенпроизводных, спиртов и эфиров сульфокислот (синтез спиртов, простых эфиров, меркаптанов, сульфидов, аминов, ненасыщенных веществ, α-окисей и других гетероциклических соединений), реакции присоединения (синтезы из α-окисей, альдольная конденсация, получение азотсодержащих производных альдегидов и кетонов, некоторые реакции присоединения по двойным и тройным связям), процессы этерификации и другие превращения кислот и их производных.
11.1. Нуклеофильное замещение
11.1.1. Механизмы нуклеофильного замещения
Некаталитическое нуклеофильное замещение может происходить в одну или две элементарные стадии. Одностадийный процесс представляет собой элементарную реакцию SN 2-замещение, где индекс 2 указывает на бимолекулярность реакции (рис.11.1).
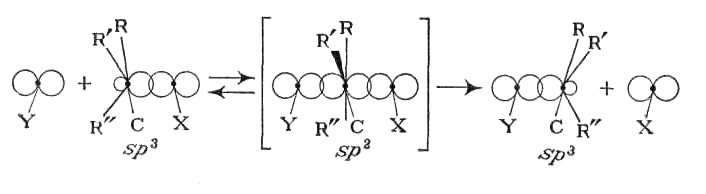
Рис. 11.1. Схема бимолекулярной реакции замещения SN 2
Как видно схемы, из приведенной на рис. 11.1, нуклеофил своей неподеленной парой р -электронов атакует углеродный атом в молекуле RR'R"CX со стороны, противоположной замещаемой группе. При образовании активированного комплекса электроны σ-связей при атакуемом атоме углерода переходят из состояния sp3- в состояние sр2 -гибридизации. В результате углеродный атом оказывается в одной плоскости с группами R, R' и R", а нуклеофил и замещаемая группа связываются с ним за счет его р -орбитали. Распад активированного комплекса в продукт RR'R"CY сопровождается обратным переходом углеродного атома в состояние sp3- гибридизации и обращением конфигурации исходного соединения, которое вызвано присоединением атома Y со стороны, противоположной разорвавшейся связи С‒X.
При двухстадийномнуклеофильном замещении разрыв связи С‒X и образование связи С‒Y происходят не одновременно. Первой элементарной реакцией является мономолекулярный гетеролитический разрыв связи С‒X с образованием карбкатиона (рис. 11.2):
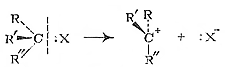
Рис. 11.2. Схема образования карбкатиона
Заряженный углеродный атом карбкатиона находится в одной плоскости с тремя σ-связями С ‒ R и с равной вероятностью атакуется нуклеофилом как со стороны отщепившейся группы X, так и с противоположной. В результате последующей бимолекулярной реакции нуклеофила с карбкатионом (вторая стадия) образуется смесь двух продуктов замещения X на Y ‒ с сохранением и с обращением конфигурации (рис. 11.3):
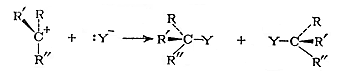
50 % 50 %
Рис. 11.3. Схема взаимодействия карбкатиона с нуклеофилом
Первая стадия ‒ мономолекулярная реакция гетеролиза ‒ идет значительно медленнее и лимитирует скорость процесса. В связи с этим двухстадийный процесс нуклеофильного замещения принято обозначать символом SN 1,где цифра 1 означает мономолекулярность определяющей стадии.
Механизмы SN 1и SN 2легко различить кинетически, если нуклеофил не является растворителем. Реакция SN 2 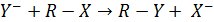 является элементарной и всегда описывается кинетическим уравнением второго порядка:
является элементарной и всегда описывается кинетическим уравнением второго порядка:

Двухстадийная реакция SN 1в общем случае описывается более сложным кинетическим уравнением. При его выводе необходимо учитывать, что реакция образования карбкатиона обратимая:

Применяя принцип стационарности Семенова-Боденштейна
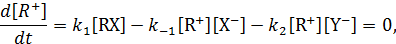
легко найти концентрацию карбкатиона и подставить ее в выражение для скорости накопления продукта, что дает: 

Из уравнения (11.2) следует, что скорость рассматриваемой реакции должна уменьшаться по мере накопления аниона Х- или при добавлении его в реакционную массу. Этот эффект получил название эффекта общего иона.
Если анион Х– обладает слабой нуклеофильностью или если к действию реагента Y– добавляется реакция с растворителем HY, сопряженным основанием которого является нуклеофил (например, НО- в воде, RO- в спирте), членом  в знаменателе уравнения (11.2) можно пренебречь и после сокращения
в знаменателе уравнения (11.2) можно пренебречь и после сокращения 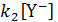 получим уравнение (11.3)
получим уравнение (11.3)

В этом случае k 1является истинной константой скорости гетеролиза по связи С‒X, и эта скорость, в отличие от скорости SN 2реакций, не зависит от концентрации нуклеофила. Такому же кинетическому уравнению соответствует SN 2 реакция, когда единственным нуклеофильным реагентом является сам растворитель, концентрация которого входит в выражение для константы скорости. В этом случае механизмы SN l и SN 2можно различить по активационным параметрам: для SN 1характерна положительная энтропия активации, для Sn 2‒ отрицательная. При SN l механизме из-за уменьшения упорядоченности системы при удлинении рвущихся связей и образовании карбкатиона энтропия возрастает. При SN 2механизме, наоборот,образование активированного комплекса из двух частиц или молекул приводит к возрастанию упорядоченности системы и следовательно к уменьшению энтропии по сравнению с исходным состоянием. Кроме того, для реакции с оптически активным соединением при SN l механизме происходит рацемизация, а при механизме SN 2 ‒ обращение оптической активности.
Механизм рассматриваемых реакций нуклеофильного замещении является в действительности более сложным. В настоящее время общепринято, что разрыв связи С‒X существенно облегчается или вообще становится возможным лишь за счет взаимодействия замещаемой группы X с электрофилами. Образование донорно-акцепторной (а) или водородной (б) связи

а б
ведет к предварительной поляризации связи С‒X и облегчает ее последующий разрыв. Замещение некоторых групп (–ОН, –OR, –SH, –SR, –NH2, –NHR, –NR2) вообще невозможно без сильного взаимодействия их с электрофилами. Например, замещение гидроксильной группы спирта при действии только бромид-аниона невозможно, но эту же реакцию легко осуществить в кислой среде. Предварительное протонирование гидроксильной группы поляризует связь С–О и делает возможным замещение этой группы анионом брома:
 Сходным путем осуществляются превращения простых эфиров, например:
Сходным путем осуществляются превращения простых эфиров, например:
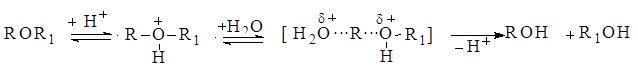
Кроме сильных протонных кислот (НС1, H2SO4) катализаторами реакций нуклеофильного замещения являются кислоты Льюиса (Ag+, Hg2+, HgCl+, HgCl2, SnCl4, А1СЦ, SbCl5, Cu2Cl2, BF3). Их действие зависит от природы отщепляемой группы. Например, протонные кислоты сильно ускоряют замещение О‒, S‒ и N‒содержащих групп и мало влияют на скорость замещения галогенов, поскольку последние являются слабыми основаниями и плохо протонируются. Атомы галогенов дают, однако, прочные донорно-акцепторные комплексы с некоторыми кислотами Льюиса, и их образование облегчает разрыв связи С‒Hal при последующей реакции замещения. Так, хлористый аллил медленно гидролизуется водой, но в присутствии однохлористой меди легко дает аллиловый спирт:
 При этом может произойти изменение механизма с SN 2в некаталитической реакции на SN 1 ‒ в каталитической:
При этом может произойти изменение механизма с SN 2в некаталитической реакции на SN 1 ‒ в каталитической:
 Ослабление С‒Х‒связи и уменьшение энергии переходного состояния в реакциях нуклеофильного замещения происходит и при образовании комплекса за счет водородной связи. Так, скорость метанолиза трифенилхлорметана описывается кинетическим уравнением третьего порядка:
Ослабление С‒Х‒связи и уменьшение энергии переходного состояния в реакциях нуклеофильного замещения происходит и при образовании комплекса за счет водородной связи. Так, скорость метанолиза трифенилхлорметана описывается кинетическим уравнением третьего порядка:
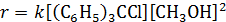
Это согласуется с механизмом, согласно которому замещение хлора происходит только при условии его специфической электрофильной сольватации другой молекулой спирта:

Образовавшийся комплекс дает далее продукт замещения:
 Такой тип механизма замещения называют многоцентровым, иногда механизмом push–push из-за того, что нуклеофил выталкивает замещаемую группу, а электрофил оттягивает (англ. pull) ее от реакционного центра.
Такой тип механизма замещения называют многоцентровым, иногда механизмом push–push из-за того, что нуклеофил выталкивает замещаемую группу, а электрофил оттягивает (англ. pull) ее от реакционного центра.
Каталитическая активность соединений, обладающих кислотными свойствами, возрастает с увеличением их кислотности. Это хорошо видно из рис. 11.4, на котором графически изображены данные по скорости сольволиза трет-бутилбромида в присутствии замещенных фенолов (0,3 моль/л).
Вещества с кислотными свойствами (вода, спирты, карбоновые кислоты и др.) часто используются как растворители для нуклеофильных реакций. Очевидно, что специфическая сольватация ими замещаемой группы ведет к ускорению процесса и возможности его проведения в умеренных условиях.
|
Рис. 11.4. Зависимость скорости реакции сольволиза трет-бутилбромида в присутствии фенолов от их кислотности Ка
|
Противоположный эффект наблюдается при специфической сольватации нуклеофила, когда образующиеся комплексы из-за рассредоточения их заряда оказываются менее реакционноспособными


Отрицательно заряженные нуклеофилы сольватируются сильнее нейтральных молекул или групп. Поэтому, например, для реакции

преобладающее влияние имеет сольватация бромид-аниона, а не атома йода в молекуле СН3I. В результате скорость этой реакции в метаноле в 58 000 раз меньше, чем в диметилформамиде, где эффект специфической сольватации отсутствует.
Аналогичная реакция с пиридином

при которой нуклеофил нейтрален, в тех же растворителях имеет близкую скорость, так как сольватация замещаемой группы компенсирует сольватацию нуклеофила.
Кроме специфической сольватации в тех случаях, когда она отсутствует или мало меняется при замене растворителя, существенное влияние на нуклеофильные реакции имеет электростатическая сольватация. Относительные полярности исходных веществ и переходного состояния неодинаковы в различных реакциях нуклеофильногозамещения (табл. 11.1), вследствие чего изменение полярности растворителя влияет на эти реакции по-разному. При большей полярности переходного состояния (реакции типа 1 и 3) скорость всегда растет из-за более сильной электростатической сольватации переходного состояния по сравнению с исходными веществами, а при обратном соотношении полярностей наблюдается уменьшение скорости (реакции типа 2 и 4).
Таблица 11.1. Влияние полярности растворителей на скорость реакций нуклеофильного замещения
| Тип реакции
| Механизм
реакции
| Влияние увеличения ε на скорость реакции
|
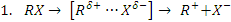
| SN 1
| Очень сильный рост
|

| SN 2
| Слабое падение
|

| SN 2
| Сильный рост
|

| SN 2
| Слабое падение
|
В качестве примера сильного влияния полярности растворителя можно привести сольволиз трет-бутилхлорида (тип 1) – при замене этанола (ε = 24) водой (ε = 81) скорость реакций возрастает в 850 раз.
На гидролиз изо-С3Н7Вг (тип 2) полярность растворителя влияет мало: скорость гидролиза уменьшается при замене этанола водой только в два раза:

Сольватация существенно влияет не только на общую скорость, но и на соотношение механизмов SN 1и SN 2. При прочих равных условиях первый из них тем более вероятен, чем выше полярность растворителя и его способность к специфической сольватации замещаемой группы и чем выше кислотность катализатора.
Это вполне понятно, так как в отсутствие перечисленных условий гетеролитическое расщепление связи С‒X связано с большой затратой энергии (>837 кДж/моль) и имело бы ничтожную скорость.
В действительности SN 1и SN 2 реакции не отделены друг от друга так резко, между ними имеется область так называемых пограничных механизмов. При «чистой» SN 2 реакции с отрицательно заряженным нуклеофилом реакционный центр в переходном состоянии имеет частичный отрицательный заряд. В зависимости от свойств растворителя, наличия катализаторов, а также от строения нуклеофила, алкильной и замещаемой групп этот заряд изменяется и может стать положительным, что сопровождается удлинением связи С‒X в переходном состоянии. Одновременно новая связь завязывается на большем расстоянии от реакционного центра (рис. 11.5). Кинетика такого пограничного механизма еще подчиняется уравнению второго порядка, однако при дальнейшем развитии рассмотренных эффектов происходит переход к чистому SN 1 механизму, когда С–Х-связь разрывается уже без участия нуклеофила.
11.1. 2. Влияние строения реагентов на нуклеофильное замещение
Влияние алкильной группы в исходной молекуле. Впереходном состоянии «чистой» SN 2 реакции углеродный атом, как мы видели, несет частичный отрицательный заряд и стабилизируется поэтом электроноакцепторными заместителями, которые способствую рассредоточению этого заряда, уменьшают энергию активированного комплекса и ускоряют реакцию. Наличие объемистых заместителей, наоборот, затрудняет подход нуклеофила и вызывает стерические эффекты, замедляющие реакцию.

а б в г
Рис. 11.5. Переходные состояния при механизме SN 2 (а), SN 1 (г) и пограничном механизме (б и в)
В реакции SN 1определяющей стадией является образована карбкатиона. По предложению Хэммонда принято считать, что активированный комплекс и образующаяся при его распаде энергетическая нестабильная частица близки по строению и энергетическому состоянию. Отсюда следует, что все факторы, стабилизирующие карбкатион, будут уменьшать и энергию активированного комплекса, ускоряя реакцию SN 1. К этим факторам относятся: наличие электронодонорных заместителей, эффекты их сопряжения и сверхсопряжения с реакционным центром. Стерическиепрепятствия при подходе реагента уже не играют роли, а присутствие объемистых заместителей даже ускоряет реакцию SN 1из-за ослабления связи С‒X.
Таким образом, ни один из рассмотренных эффектов заместителей не действует одинаково на реакции SN 1 и SN 2. Например, накопление метальных групп у реакционного центра в ряду алкилгалогенидов облегчает образование карбкатиона и ускоряет реакцию SN 1из-за сверхсопряжения метильных групп с реакционным центром и их стерического и индуктивного эффекта:
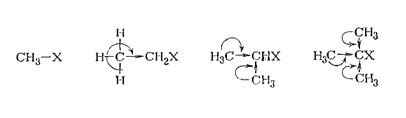
Скорость реакции SN 2, наоборот, возрастает с уменьшением числа метильных групп из-за уменьшения стерических препятствий и снижения положительного индуктивного влияния заместителей. В результате первичные алкилгалогениды реагируют по SN 2 механизму, а третичные - по SN 1.
Между этими предельными случаями находится область пограничных механизмов, когда из-за изменения заряда на реакционном центре влияние заместителей становится обратным и сходным с реакцией SN 1. По этой причине в зависимости от донорно-акцепторных свойств заместителей, связанных с реакционным центром, наблюдается понижение или повышение скорости с прохождением ее через минимум (рис. 11.6). Так, скорости алкоголиза СН3Вг, С2Н5Вr, изо-С3Н7Вr и тpeт-C4H9Br этанолом относятся как 1: 0,40: 0,68: 2900. Очевидно, что структурные влияния являются еще одним способом установления механизма нуклеофильного замещения.

|
Рис. 11.6. Влияние электронодонорного эффекта заместителей на скорость реакции нуклеофильного замещения
|
Аналогичное, но значительно более быстрое изменение механизма процесса имеет место в ряду СН3‒CI PhCH2‒CI Ph2CH‒CI Ph3C‒С1.
Здесь гидролиз по механизму SN l наблюдается уже, начиная со второго соединения. Причиной столь сильного облегчения ионизации и связанного с этим более быстрого перехода к механизму SN l является значительная стабилизация карбкатиона за счет эффекта сопряжения и делокализации положительного заряда с участием π-электронных орбиталей бензольного кольца:
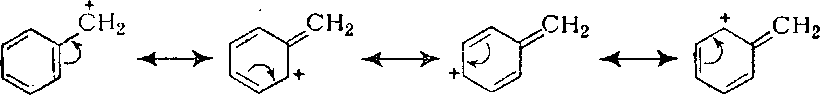
В отношении реакционной способности бензилхлорид аналогичен трет-бутилхлориду. При переходе к Ph2CHCl и к Ph3CCl эффект становится все более отчетливо выраженным и атака по механизму SN l облегчается еще сильнее, поскольку возможность делокализации положительного заряда для карбкатионов, образующихся из этих галогенпроизводных, соответственно возрастает.
Группа СН2 в бензилхлориде также атакуется значительно легче по механизму SN 2, чем группа СН2 в ионном соединении, например в МеСН2С1 (замещение идет приблизительно в 100 раз быстрее), поскольку sp 2‒гибридизованный атом углерода в переходном состоянии SN 2 может использовать свою негибридизованную р‒орбиталь для взаимодействия не только с нуклеофилом и уходящим ионом С1-, но также с π-орбиталью фенильной группы, которая при этом дополнительно стабилизуется.
Аналогичная стабилизация карбкатионов наблюдается и для аллилгалогенидов:

В этом случае атака по механизму SN l облегчена и аллилгалогениды подобно бензилгалогенидам обладают обычно значительно большей реакционной способностью по сравнению, например, с СН3СН2СН2С1 и PhCH2CH2CH2Cl, для которых стабилизация карбкатионов за счет делокализации положительного заряда невозможна. Атака по механизму SN 2 сильно ускоряется также в результате взаимодействия атакуемого атома углерода с π-орбиталью двойной связи в переходном состоянии, аналогично тому, как это имеет место в рассмотренном выше случае замещения в бензилхлориде.
Что же касается винилхлоридов, таких как, например, СН2 = СНС1, а также галогенбензолов, то они очень устойчивы по отношению к нуклеофилам. Это объясняется тем, что атом галогена в этих соединениях связан с атомом углерода, имеющим sp 2‒гибридизацию, вследствие чего электронная пара связи С‒С1 оттянута ближе к углероду по сравнению со связями, включающими sp 3‒гибридизованиые атомы углерода.
Смещение электронной плотности от атома хлора в сторону двойной связи, благодаря эффекту сопряжения (+ M эффект), действует одновременно с сильным электроноакцепторным индуктивным эффектом атома хлора (− I эффект). Однако − I > + M, поэтому галоген несёт на себе небольшой отрицательный заряд, а частичный положительный заряд на атоме углерода уменьшается по сравнению хлорэтаном. 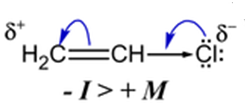 В этих соединениях связь С‒С1 более прочная, чем в случае, например, СН3СН2С1, и дипольный момент таких связей меньше (табл.11.2).
В этих соединениях связь С‒С1 более прочная, чем в случае, например, СН3СН2С1, и дипольный момент таких связей меньше (табл.11.2).
Таблица 11.2. Сравнительные значения длин и энергии связей, а также дипольных моментов в молекуле винилхлорида и некоторых хлоралканов
| Соединение
| Энергия связи C-Cl, кДж/моль
| Длина связи C-Cl, нм
| Дипольный момент, 10−30 Кл·м
|
| CH2=CHCl
| 374,89
| 0,169
| 4,80
|
| CH3-CH2Cl
| 336,39
| 0,179
| 6,66
|
| CH3Cl
| 349,78
| 0,176
| 6,19
|
Вследствие этого тенденция к ионизации (SN l) выражена для винилхлоридов слабее, а положительный заряд на атоме углерода недостаточен для того, чтобы этот атом мог эффективно атаковаться нуклеофилом (SN 2). Кроме того, π-электроны двойной связи в этих соединениях препятствуют приближению атакующего нуклеофила. Наличие двойной связи в этих условиях не должно способствовать стабилизации ни переходного состояния, возникающего по механизму SN 2, ни карбкатиона, образующегося при механизме SN 1.
Данные выводы справедливы также и для галогенбензолов с sp 2‒гибридизованными углеродами и π-орбитальными системами ароматического кольца.
Кислородные заместители в алкильной группе имеют разное влияние. Например, α-хлорэфиры ввиду возможности сопряжения с реакционным центром реагируют очень быстро по SN1- механизму
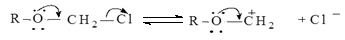
Наоборот, в β-хлорэфирах сопряжение невозможно, и их реакционная способность меньше, чем у незамещенного хлорпроизводного.
На скорость реакций иногда сильно влияет непосредственное взаимодействие одного из атомов заместителя с относительно далеко расположенным от него реакционным центром. При нуклеофильном замещении в галогенкарбоновых кислотах этот эффект соседних групп проявляется очень ярко. Их сольволиз в кислых средах ускоряется, когда галоген и карбоксильная группа расположены таким образом, что образуется внутримолекулярная водородная связь с замещаемой группой, например в α-галогенкарбоновых кислотах:
 В щелочной среде, когда кислота превращается в соль, такое электрофильное активирование реакции невозможно, но зато появляется эффект сопряжения, который способствует расщеплению С‒Х-связи с внутримолекулярной атакой реакционного центра и образованием нестабильного лактона, быстро переходящего в продукт реакции:
В щелочной среде, когда кислота превращается в соль, такое электрофильное активирование реакции невозможно, но зато появляется эффект сопряжения, который способствует расщеплению С‒Х-связи с внутримолекулярной атакой реакционного центра и образованием нестабильного лактона, быстро переходящего в продукт реакции:

Влияние замещаемой группы. При прочих равных условиях нуклеофильное замещение как по SN 1, так и по SN 2 механизму должно протекать тем быстрее, чем ниже энергия гетеролитического разрыва связи С‒X. Усредненная относительная способность к замещению для некоторых замещаемых атомов и групп такова:
| F-
| Cl-
| Br-
| I-
| C6H5SO2O-
| H2O+-
|
| 10-4
|
|
|
|
|
|
Наиболее легко отщепляемыми группами являются обычно анионы сильных кислот, например С6Н4SO3-. Это во многом связано со стабилизацией образующегося аниона за счет его лучшей поляризуемости или эффектов сопряжения, например, для эфиров сульфокислот и диалкилсульфатов:

Кислые эфиры серной кислоты обладают лишь слабой алкилирующей способностью, так как у них легче разрывается связь S‒О.
Чем выше основность отщепляемой группы, тем труднее она отщепляется атакуемым нуклеофилом. Такие сильно основные группы, как, например, OR, ОН, связанные с углеродом через небольшие трудно поляризуемые атомы кислорода, в обычных условиях, как правило, не способны к замещению. Однако, в кислой среде они все же могут замещаться вследствие начальной протонизации, приводящей к образованию положительно заряженных ониевых соединений (а не нейтральных молекул), атакуемых нуклеофилом. В результате замещение менее основной группировки может идти легче:

Галогенпроизводные характеризуются высокой химической активностью, объясняющейся поляризацией связи С‒Hal. На атоме углерода образуется частичный положительный заряд, на атоме галогена – частичный отрицательный заряд. Таким образом, атомы галогенов проявляют отрицательный индуктивный эффект по отношению к связанному с ним атому углерода. Этот эффект передается по цепи простых связей с сильным затуханием.
Реакционная способность галогенпроизводных зависит от природы галогена. На подвижность атома галогена оказывают влияние полярность связи и поляризуемость этой связи. Полярность связи углерод – галоген определяется электроотрицательностью атома галогена и длиной связи. Степень полярности связи можно оценить величиной дипольного момента связи. Для связей С-Hal дипольные моменты (μ) имеют следующие значения:
| Связь
| С-F
| С-Cl
| С-Br
| С-I
|
| μ, Д
| 1,83
| 2,05
| 2,04
| 1,8
|
Как видно из сравнения величины дипольных моментов, полярность связи тем выше, чем больше электроотрицательность галогена. Нарушение закономерности для связей С-F и С-Cl объясняется сильным увеличением длины связи при переходе от элемента второго периода к элементу третьего периода. Таким образом, казалось бы, в реакции замещения должны вступать наиболее легко алкилгалогениды с большей полярностью связи. На самом же деле порядок подвижности галогенов в галогенуглеводородах обратный: I > Br > Cl > F.
Такая закономерность объясняется более легкой поляризуемостью связи C-I по сравнению с остальными галогенами. Поляризуемость - это способность к дополнительной поляризации под влиянием внешнего электрического поля. Поляризуемость тем выше, чем дальше от ядра атома находятся электроны, образующие связь. Все вышесказанное находит выражение и в величинах энергии разрыва связей (в кДж/моль):
| Группа Х
| F
| Cl
| Br
| I
|
| CH3СH2X
| 460,5
| 336,6
| 272,1
| 221,9
|
| CH2=CHX
| 489,8
| 372,6
| 309,8
| -
|
При прочих равных условиях, чем больше относительная способность к замещению, тем выше роль пограничных или SN 1 механизмов в реакциях нуклеофильного замещения.
Влияние нуклеофильного реагента. Факторы, влияющие на скорость реакций нуклеофильного замещения и связанные со строением нуклеофильного реагента, объединяют понятием нуклеофильность. К этим факторам относятся основность и поляризуемость нуклеофила.
Связь нуклеофильности с основностью обусловлена аналогией протолитических реакций и реакций нуклеофильного замещения: в протолитической реакции основание за счет своей свободной электронной пары присоединяет протон; в реакции нуклеофильного замещения этой же электронной парой осуществляется атака на углеродный атом, имеющий дефицит электронов. В тех случаях, когда другие факторы, влияющие на нуклеофильность, остаются неизменными, наблюдается корреляция между константой скорости реакции SN 2-замещения (нуклеофильностью) и константой основности нуклеофильного реагента (основностью). Количественно эта зависимость выражается уравнением Брёнстеда:
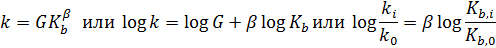
где k ‒ константа скорости, Кb ‒ константа основности нуклеофила, G и β ‒ эмпирические константы.
Чем больше β, тем быстрее возрастает скорость с увеличением основности нуклеофила, что характерно для типичных SN 2 реакций, в которых нуклеофил Y в большой степени связывается с углеродным атомом уже в переходном состоянии (Y--C--X). По мере перехода от SN 2 к SN 1 механизму завязывание новой связи происходит на все большем расстоянии (Y----С----X), и величина β уменьшается.
 На рис. 11.7 эта зависимость показана для изменения относительных скоростей алкоголиза (k1/k0) некоторых хлоридов
На рис. 11.7 эта зависимость показана для изменения относительных скоростей алкоголиза (k1/k0) некоторых хлоридов

в зависимости oт относительной основности алкоголятов (Кb,i/Kb,0).
Трет -бутилхлорид реагирует по SN 1 механизму, и относительная реакционная способность алкоголятов в этой реакции (кривая 3) не зависит от их основности (β≈ 0).
| Рис. 11.7. Зависимость относительной реакционной способности алкоголятов от их основности для реакций с органическими хлоридами:
1 – с н -бутилхлоридом;
2 – с аллилхлоридом;
3 – с трет -бутилхлоридом
|
Кроме основности на нуклеофильность сильно влияет поляризуемость реакционного центра нуклеофила. В объемистых ионах (S
–,I
–, Вr
–) или при наличии в анионе кратных связей (N
3–, CN
–) электроны легко смещаются в направлении положительно заряженного атома углерода в атакуемой молекуле. Поляризованная молекула нуклеофила завязывает с ним связь на большем расстоянии и реагирует значительно быстрее нуклеофилов той же или даже большей основности, но неспособных к поляризации. Например, усредненная относительная нуклеофильность легко поляризуемых иона C
6H
5S
– в 470 раз выше, чем для иона С
2Н
5О
–, тогда как основность иона С
2Н
5О
– (
рКb = –2) на много порядков больше, чем для иона C
6H
5S
– (рКb = 4,6). Высокой нуклеофильностью обладают также ионы HS
– и CN
–, молекулы аммиака и аминов. В результате условия синтеза меркаптанов, сульфидов, аминов, нитрилов нередко оказываются даже более мягкими, чем условия гидролиза алкилхлоридов щелочами.
Эдвардс предложил следующее уравнение для количественной оценки нуклеофильности EN:
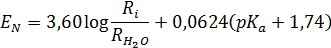
где R ‒ рефракция (поляризуемость), а рКа ‒ кислотность кислоты, сопряженной нуклеофилу.
Стандартом служит вода, для которой нуклеофильность принята равной нулю. Значения EN для некоторых нуклеофилов приведены в табл. 11.2.
Таблица 11.2. Значение нуклеофильности EN нуклеофилов
| Нуклеофил
| EN
| Нуклеофил
| EN
|
| F–
| -0,23
| HO–
| 1,60
|
| H2O
|
| NH3
| 1,84
|
| CH3COO–
| 0,95
| CN–
| 2,01
|
| Cl–
| 1,21
| I–
| 2,02
|
| C6H5O–
| 1,46
| CH3O–
| 2,74
|
| Br–
| 1,57
| C2H5O–
| 3,78
|
Для аминов нуклеофильность изменяется в ряду:
R2NH ≈ RNH2 >> NH3.
 Рис. 11.8. Зависимость скорости реакции замещения от нуклеофильности реагентов
Рис. 11.8. Зависимость скорости реакции замещения от нуклеофильности реагентов
| При прочих равных условиях чем сильнее нуклеофильность реагента, тем более вероятно протекание реакции по SN 2 механизму. Наоборот, со слабыми нуклеофилами более быстрой может оказаться реакция SN 1, и тогда процесс пойдет по этому пути. Такая зависимость с обращением механизмов действительно наблюдается, причем в области SN 1 механизма скорость реакции не зависит от природы нуклеофила (рис. 11.8).
|
При обмене галогенов реакция SN 2всегда протекает в сторону замещения более электроотрицательного атома на более нуклеофильный (F и С1 на Вг и I). Однако при SN l замещении образуется продукт с более электроотрицательным заместителем (т. е. RF из RC1). Для изменения механизма используют соли или катализаторы с сильно электрофильным катионом (AgF, HgF2, SbF3, SbF5). Например, при использовании фторида сурьмы (V) реакция замещения протекает по механизму:

Ассоциация отрицательно заряженного нуклеофила с катионом с образованием ионной пары уменьшает реакционную способность нуклеофила, как и электрофильная сольватация за счет водородных связей.

В средах с низкой диэлектрической проницаемостью (ε< 40) заряженные нуклеофилы находятся в основном в виде ионных пар. Особенно прочные ионные пары образуются ионами небольшого размера, поэтому увеличение размеров катиона всегда способствует росту нуклеофильности аниона (нуклеофильная реакционная способность LiCl обычно в 10-1000 раз меньше, чем для хлоридов тетраалкиламмония R4N+ Cl–). Диссоциация ионных пар облегчается также в результате специфической сольватации катионов, например,

Такая сольватация оказывает влияние, аналогичное увеличению размеров катиона, и сильно ускоряет нуклеофильную реакцию с участием этой ионной пары.
Анализ кинетических данных по влиянию природы нуклеофила и замещаемой группы на скорость реакций нуклеофильного замещения показывает, что некоторые легко поляризуемые нуклеофилы (например, I– и Вr–) являются в то же время легко замещаемыми группами. Это позволяет использовать их в качестве нуклеофильных катализаторов. Например, анион йода сильно ускоряет относительно медленные реакции гидролиза алкилхлоридов водой. Механизм нуклеофильного катализа в данной реакции состоит в предварительном замещении хлор‒аниона йодом и в последующем гидролизе образовавшегося алкил- иодида с образованием спирта и регенерацией йод‒аниона:

Конкуренция нуклеофилов при замещении. При проведении реакций нуклеофильного замещения в реакционных смесях обычно присутствуют несколько нуклеофилов, что обусловливает образование побочных веществ. Кроме того, некоторые нуклеофилы обладают двумя реакционными центрами и дают разные продукты замещения. При SN 2 реакции органическое вещество преимущественно реагирует, как мы видели выше, с реагентом, имеющим наибольшую нуклеофильность, зависящую от основности и поляризуемости. В отличие от этого, при SN l замещении карбкатион ввиду его высокой реакционной способности имеет меньшую избирательность, но все же предпочтительно взаимодействует с нуклеофилом, имеющим наибольшую электронную плотность (или электроотрицательность). Это правило Корнблюма имеет болшое значение для предсказания необходимых условий реакции.
Если взять для примера реакции с нуклеофилами, обладающими двойственной реакционной способностью (C=N, О‒N‒О и др.), то они дают обычно смесь продуктов замещения (нитрилы и изонитрилы, нитросоединения и нитриты) в зависимости от электроотрицательности и поляризуемости атомов:
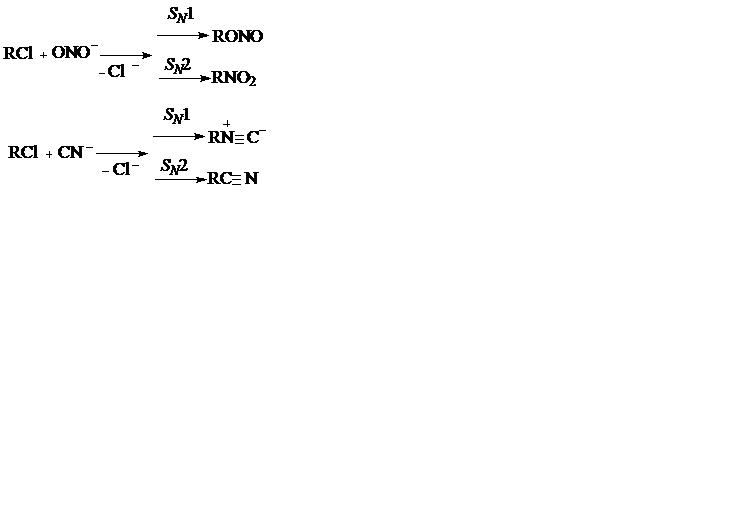
При нуклеофильном замещении нередко происходит изомеризация, наиболее характерная для механизмов SN 1. Ее причина состоит в том, что образующийся карбкатион, имеющий только секстет электронов, перед тем как он прореагирует с соответствующим нуклеофилом, способен к внутримолекулярной стабилизации за счет миграции к нему отдельных атомов или групп. В результате получается иной карбкатион (или несколько ионов), который при последующей реакции с нуклеофилом дает смесь продуктов замещения и отщеплени