Билет 1. СПИРТЫ: определение, изомерия, номенклатура
Спиртами называют производные углеводородов, содержащие группу –ОН, называемую гидроксильной группой или гидроксилом. По числу гидроксильных групп, содержащихся в молекуле, спирты делятся на одноатомные, двухатомные, трехатомные и т.д.
Предельные одноатомные спирты
Предельные одноатомные спирты имеют общую формулу R–OH. В зависимости от того, при каком углеводородном атоме находится гидроксил, различают первичные, вторичные и третичные спирты:

1-пропанол 2-бутанол 2-метил-2-пропанол
Изомерия одноатомных предельных спиртов определяется строением углеродного скелета и положением гидроксильной группы в цепи.
В номенклатуре ИЮПАК наличие гидроксильной группы обозначается суффиксом «ол» с указанием ее положения в цепи цифрой. При нумерации цепи гидроксил имеет предпочтение перед радикалом, кратной и тройной связью, галогеном.
Простейшие спирты называют по наименованию радикалов, с которым соединена гидроксильная группа:
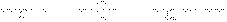
изопропиловый спирт трет-бутиловый спирт изоамиловый спирт
В рациональной номенклатуре спирты называют, как производные простейшего спирта CH3─OH – карбинола

1-пропанол (этилкарбинол) 2-бутанол (метилэтилкарбинол)
Способы получения
1) Гидролиз галогеналкилов при нагревании с водой или водным раствором щелочи позволяет получить спирты различного строения. Легче всего гидролизуются йодпроизводные, труднее всего–фторпроизводные. Атомы галогена, связанные с третичным углеродом, гидролизуются значительно легче, чем при вторичном и первичном атоме углерода:

2) Гидратация алкенов (высокое давление и температура, катализатор) позволяет получать помимо этанола вторичные и третичные спирты:

этилен этиловый спирт
3) Восстановление альдегидов и сложных эфиров в присутствии катализаторов приводит к образованию первичных спиртов, а кетонов – вторичных спиртов (катализатор никель):

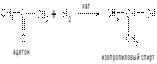
4) Действие металлорганических соединений на альдегиды и кетоны используется для получения спиртов в лабораторной практике:

5) Ферментативное сбраживание углеводов.
Глюкозосодержащий раствор под влиянием ферментов дрожжей превращается в бражку, содержащую до 10% этанола, который отгоняют и очищают. Попутно с этанолом образуются первичные амиловый, бутиловый и пропиловый спирты. Смесь этих спиртов называется сивушным маслом. C6H12O6  2C2H5OH + 2CO2
2C2H5OH + 2CO2
Химические свойства
Спирты не обладают ярко выраженным кислым или основным характером. Спирты и их водные растворы не проводят электрический ток. Способность к диссоциации у спиртов меньше, чем у воды, т. к. органический радикал является донором электронной плотности. Спирты обладают высокой реакционной способностью. Реакции, в которые вступают спирты, можно разделить на несколько групп:
А) Реакции, идущие с участием атома водорода гидроксильной группы.
1) Замещение атома водорода в гидроксиле на металл. В этой реакции спирт проявляет свойства слабой кислоты. Реакция протекает под действие щелочных металлов и магнийгалогеналкилов. Продукты реакции называются алкоголятами:
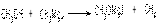
метанол иодистый метилатмагний метан
метилмагний иодид

2) Замещение атома водорода на ацильный радикал с образованием сложных эфиров. Эта реакция называется реакцией этерификации:

уксусная кислота метанол метилацетат
Б) Реакции, идущие с участием гидроксильной группы
1) Замещение гидроксильной группы на галоген. Эту реакцию обычно проводят, действуя галогенидами фосфора или серы или галогеноводородными кислотами в присутствии водоотнимающих агентов:

этанол тионил хлорид хлорэтан
2)Спирты способны отщеплять воду с образованием олефинов. Эта реакция называется реакцией внутримолекулярной дегидратации. Обычно ее проводят в присутствии избытка H2SO4 при нагревании:

этанол этилен
3)Межмолекулярная дегидратация будет наблюдаться при избытке спирта и будет протекать параллельно внутримолекулярной дегидратации:

4) В промышленности возможна замена гидроксильной группы на аминогруппу. Эта реакция протекает при действии аммиака на спирты при температуре 3000С и в присутствии катализатора Al2O3:
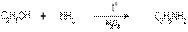
этанол этиламин
В) Реакции окисления.1) Спирты при пропускании их паров над мелкораздробленной медью подвергаются дегидрированию с образованием альдегидов и кетонов. Это характерно только для первичных и вторичных спиртов:

2) Спирты окисляются в присутствии сильных окислителей. При окислении спиртов действие окислителя направлено на атом углерода, содержащий гидроксильную группу. Первичные и вторичные спирты дают соответствующие карбонилы. Для окисления третичных спиртов необходимы более жесткие условия, при этом происходит разрыв С-С связей:

этанол

Многоатомные спирты
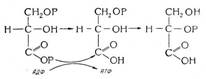
Представителями многоатомных спиртов являются гликоли (диолы) – двухатомные спирты и глицерины (триолы) – трехатомные спирты. В соответствии с правилом Эльтекова, в многоатомных спиртах гидроксильные группы располагаются у разных углеродных атомов. (Атом углерода способен удержать только одну гидроксильную группу).
Изомерия моногоатомных спиртов определяется строением углеродного скелета, а также расположением в цепи и взаимным расположением гидроксилов.
Для обозначения многоатомных спиртов применяют тривиальные названия (глицерин, инозит), а также рациональную номенклатуру и ИЮПАК.
По рациональной номенклатуре двухатомные спирты называют по названию этиленового углеводорода с добавлением слова гликоль.
Например:
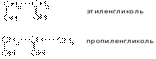
В номенклатуре ИЮПАК для многоатомных спиртов сохраняются те же принципы, что и для одноатомных.
Способы получения
Многоатомные спирты можно получит теми же методами, что и одноатомные.
Среди специфических методов получения многоатомных спиртов, наиболее часто используются следующие:
1) Гидратация α-оксидов
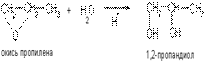
2) Окисление олефинов пероксидом водорода или разбавленным раствором перманганата калия:
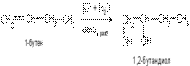
3) Синтез глицерина из пропилена осуществляется по схеме:
 По другому методу аллиловый спирт обрабатывают пероксидом водорода:
По другому методу аллиловый спирт обрабатывают пероксидом водорода:
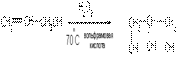
4) Гидролиз жиров
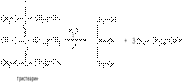
1) В отличие от одноатомных спиртов, многоатомные спирты проявляют более выраженный кислый характер. Они способны давать полные и неполные алкоголяты (гликоляты, глицераты) не только со щелочными металлами, но и с оксидами других металлов. Реакция с гидроксидом меди является качественной реакцией для обнаружения многоатомных спиртов:

2) С минеральными и органическими кислотами многоатомные спирты образуют полные и неполные эфиры:


Способы получения
1) Значительные количества фенола получают сплавлением натриевой соли бензолсульфокислоты со щелочами (реакция нуклеофильного замещения)

2) Гидролиз галогенопроизводных (реакция Ульмана). В технике получают фенол с хорошим выходом из хлорбензола

3) Разложение гидропероксидов. В промышленности фенол совместно с ацетоном получают при разложении гидроперекиси кумола
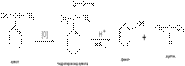
4) Окислительное декарбоксилирование карбоновых кислот. Реакция протекает в присутствии солей меди(II) при температуре 200–3000С. В реактор пропускают водяной пар и воздух:

5) Разложение солей диазония в присутствии раствора серной кислоты:
Химические свойства
По своему строению фенолы аналогичны спиртам. Однако гидроксильная группа и непосредственно связанное с ней бензольное кольцо оказывают сильное влияние друг на друга, что обуславливает специфичность свойств фенолов.
1) Кислые свойства фенолов более ярко выражены, чем у спиртов. Это объясняется тем, что свободная электронная пара атома кислорода в феноле оттянута к ядру (+М–эффект).

 Многоатомные фенолы обнаруживают еще более сильные кислые свойства, чем фенол. Например, для пирокатехина характерно выпадение нерастворимой свинцовой соли при его прибавлении к раствору ацетата свинца:
Многоатомные фенолы обнаруживают еще более сильные кислые свойства, чем фенол. Например, для пирокатехина характерно выпадение нерастворимой свинцовой соли при его прибавлении к раствору ацетата свинца:
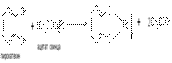
Феноляты железа имеют комплексный характер. Их растворы интенсивно окрашены. Эта реакция используется для качественного открытия фенолятов. Фенол дает с раствором FeCl3 фиолетовую окраску, крезолы – голубую, пирокатехин – зеленую.
2) Фенолы способны образовывать простые и сложные эфиры. При действии на феноляты галогеналкилов, галогенангидридов и ангидридов кислот:


3) Гидроксильная группа оказывает очень большое влияние на ароматическое кольцо, увеличивая его реакционную способность в о- и п- положениях, поэтому фенол очень легко вступает во всевозможные реакции электрофильного замещения и конденсации:
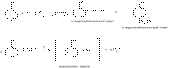
Реакция конденсации с алифатическими альдегидами имеет большое практическое значение, так как лежит в основе производства пластических масс и лаковых основ.
4) При гидрировании водородом в присутствии катализаторов фенолы превращаются в спирты ряда циклогексана:

5) Вступление гидроксильной группы в ядро делает его менее устойчивым к действию окислителей. Наиболее легко окисляются моногоатомные фенолы, содержащие гидроксильные группы в о- и п- положениях:

6) Из всех фенолов флороглюцин наиболее склонен к таутометрии. Например с гидроксиламином он дает триоксим.
Способы получения
1. Замещение атомов водорода в углеводородах на галоген
а) галогенирование предельных углеводородов протекает по радикальному механизму при облучении:

Замещение может идти и дальше.
Фтористые алкилы получают обычно действием фтористого серебра на йодистые алкилы:

или действием на алканы CoF3 – последний при 200 оС медленно выделяет F2:
2CoF3  → 2CoF2 + F2
→ 2CoF2 + F2
CH3-CН3 + 12CоF3 → CF3-CF3 + 6HF + 12 CоF2
б) бензол и его гомологи галогенируются легче, чем алканы. Причем на холоде и в присутствии катализатора (хлоридов тяжелых металлов) галоген замещает атомы водорода в бензольном ядре, а при нагревании или на свету в отсутствии катализатора замещаются атомы водорода в боковой цепи:

в). При высоких температурах (400 – 600 оС) удается заместить атомы водорода в алкенах, не нарушая кратной связи:

2. Присоединение галогенов или галогеноводородов к ненасыщенным углеводородам:

3. Получение галогенопроизводных из спиртов. Чаще всего на спирты действуют галогенидами фосфора, серы, а также галогеноводородными кислотами в присутствии водоотнимающих средств:

4. Общий метод синтеза дигалогенопроизводных – это действие пентахлорида или пентабромида фосфора на альдегиды и кетоны:

При этом получаются соединения с двумя атомами галогена при одном атоме углерода.
5. Хлороформ, бромоформ, йодоформ можно получить действием соответствующего галогена в щелочном растворе на ацетальдегид

Физические свойства
Низшие алкилгалогениды – газообразные вещества, средние – жидкости, высшие – твердые тела. Физические свойства галогенопроизводных зависят от строения радикала, вида и количества атомов галогена в молекуле. С увеличением длины углеродной цепочки и с переходом от фтора к йоду температура кипения и плотность моногалогенопроизводных возрастает. Дигалогенопроизводные – тяжелые масла или твердые вещества. Все галогенопроизводные практически нерастворимы в воде, хорошо растворимы в органических растворителях, большинство имеют специфический, часто резкий запах, раздражающий слизистую оболочку, некоторые обладают анестезирующим действием (СН2Сl2, СНСl3), токсичны, являются антисептиками (СНJ3).
Химические свойства
Большинство галогенопроизводных углеводородов - весьма реакционноспособные соединения, широко применяемые в разнообразных синтезах. Это определяется тем, что атом любого галогена обладает большим сродством к электрону, чем атом углерода. σ-Связь C─Halg в галогеналкилах сильно поляризована, что приводит к возникновению пониженной электронной плотности на атоме углерода и повышенной на атоме галогена. Для органических молекул характерна так называемая поляризуемость, т.е. cпособность увеличивать полярность связи при подходе атакующего реагента. Поляризуемость связи тем больше, чем более объемиста и подвижна электронная оболочка атомов, образующих связь. Так, если полярность связей C-F, C-Cl, C-Br и C-J довольно близка, то поляризуемость связи C-J значительно больше, чем связи C-F. В точном соответствии с поляризуемостью во всех реакциях нуклеофильного замещения иодпроизводные максимально активны, а фторопроизводные практически не реакционноспособны.
На реакционную способность галогена оказывает большое влияние, и строение органического радикала. При нахождении галогена у углерода с кратной связью свободная электронная пара атома галогена сопряжена с π-электронами двойной связи:

Это проявляется в заметном укорачивании связи C–Cl от 0,176 до 0,169 нм вследствие дополнительного взаимодействия и в очень сильном уменьшении реакционной способности. Атомы галогена при двойной связи практически не реакционноспособны. Напротив, у атома галогена в аллильном положении реакционная способность резко увеличивается. Это объясняется тем, что способность таких соединений к диссоциации заметно повышена

Образующийся аллильный карбкатион стабилизируется вследствие смещения π-электронной плотности на положительно заряженный атом углерода. Поэтому образование такого карбкатиона энергетически значительно более выгодно, чем алкилкатиона. Все реакции замещения аллильного галогена идут по механизму SN1 и их скорость на несколько порядков выше, чем для алкилгалогенидов. По той же причине, что и в хлористом виниле, атом галогена в бензольном ядре мало реакционноспособен, однако бензильный атом галогена, как и в бромистом аллиле, очень легко вступает в реакции нуклеофильного замещения.
Реакция гидролиза
При действии свежеприготовленной гидроокиси серебра или воды в присутствии щелочей галогеналкилы дают спирты

Для первичных и вторичных алкилгалогенидов механизм этих реакций SN2

Отрицательно заряженная гидроксильная группа атакует положительно заряженный атом углерода со стороны, противоположной отрицательно заряженному атому брома. При наличии достаточной энергии гидроксил приближается настолько, что между ним и атомом углерода начинает образовываться связь, а связь между атомами углерода и брома начинает разрываться. В этом переходном состоянии атом углерода и все три атома водорода находятся в одной плоскости (молекула «уплощена»). Затем анион брома выталкивается и образуется молекула метилового спирта. Такой процесс называется реакцией нуклеофильного замещения второго порядка (SN2): нуклеофильного потому, что атакующая частица заряжена отрицательно; второго порядка - потому что скорость реакции зависит от концентрации как бромистого метила, так и гидроксила.
У третичных алкилгалогенидов подход отрицательно заряженной частицы затруднен имеющимися объемными заместителями, и процесс идет по другому механизму:
Хотя и в очень малой степени, но все же происходит процесс диссоциации третичного бромистого бутила. Образующийся третичный бутилкатион мгновенно реагирует с находящимися в растворе нуклеофильными частицами:

В этом случае скорость реакции зависит только от процесса диссоциации и, следовательно, от концентрации в реакционной смеси третичного бромистого бутила и реакция в целом называется реакцией нуклеофильного замещения первого порядка SN1.
Реакции замещения
К реакциям замещения, протекающим по нуклеофильному механизму, относятся реакции замещения галогена на CN-группу, аминогруппу, нитрогруппу, водород и т. д.


Реакция β-элиминирования (отщепления).
При действии на алкилгалогениды спиртовыми растворами щелочей образуются алкены
Получение магнийорганических соединений.
Способы получения
Химические свойства
1) Эфиры, в отличие от спиртов, с металлическим натрием реагируют только при нагревании и без выделения водорода:
2) Концентрированные йодистоводородная и серная кислоты разлагают простые эфиры:
3) Эфиры растворяются в концентрированных протонных кислотах (HCl, HBr) с выделением тепла. При этом образуются непрочные соединения солеобразного характера – оксониевые соединеия.
Образующиеся комплексные соли напоминают соли аммония.
4) При хранении простые эфиры медленно окисляются кислородом воздуха, образуя крайне взрывчатые гидропероксиды:
Эфиры перед использованием всегда необходимо проверять на присутствие перекиси реакцией с KI в присутствии крахмала.
Способы получения
Альдегиды и кетоны получают рядом общих методов.
1. Дегидрирование или окисление спиртов. В этом случае из первичных спиртов образуются альдегиды.
2. Альдегиды получают пиролизом кислот и их смесей в виде паров над оксидами марганца, тория, кальция, цинка (MnO2, ThO2, CaO, ZnO) при 400–450оС:
альдегиды и кетоны могут быть получены пиролизом кальциевых и бариевых солей карбоновых кислот. Эта реакция дает очень низкий выход.
3.Гидролиз геминальных (галогены находятся у одного углеродного атома) галогенопроизводных:
4. Гидратацией ацетилена и его гомологов в условиях реакции Кучерова:
5. В технике альдегиды получают прямым присоединением оксида углерода (II) и водорода к олефинам при 100–2000С под давлением 10 – 20 МПа в присутствии кобальтового или никелевого катализаторов.Эта реакция называется оксосинтезом:
6. Альдегиды можно получать восстановлением хлорангидридов кислот водородом в присутствии палладия (реакция Роземунда)
Способы получения
кетоны получают рядом общих методов.
1. Дегидрирование или окисление спиртов вторичных – кетоны:
2. кетоны получают пиролизом кислот и их смесей в виде паров над оксидами марганца, тория, кальция, цинка (MnO2, ThO2, CaO, ZnO) при 400–450оС:
Кетоны могут быть получены пиролизом кальциевых и бариевых солей карбоновых кислот. Эта реакция дает очень низкий выход.
3. Гидратацией ацетилена и его гомологов в условиях реакции Кучерова:
4.Ароматические кетоны могут быть получены по реакции Фриделя-Крафта из ароматических углеводородов и хлорангидридов кислот:
Химические свойства
Карбонильные соединения отличаются большой реакционной способностью. Большинство их реакции обусловлено присутствием активной карбонильной группы. Двойная связь карбонильной группы сходна по физической природе с двойной связью между двумя атомами углерода (σ–связь + π–связь). С другой стороны, кислород является более электроотрицательным элементом, чем углерод, и поэтому электронная плотность вблизи атома кислорода больше, чем вблизи атома углерода.
Реакции окисления
1.Кетоны плохо окисляются кислородом воздуха, слабыми и сильными окислителями до кислот с тем же числом углеродных атомов
2.Окисление кетонов протекает с разрывом углеродной цепочки в разных направлениях в зависимости от строения кетонов. Разрыв углеродной цепи требует действий сильных окислителей и жестких условий реакции:
Реакции конденсации
1. Альдольная конденсация
Альдегиды и метилкетоны в слабоосновной среде (ацетат калия, поташ) подвергаются альдольной конденсации. Альдольная конденсация идет только за счет группы, находящейся в α-положении к карбонилу, так как только водородные атомы этой группы в достаточной степени активизируются карбонильной группой:
В результате реакции конденсации образуются новые углерод-углеродные связи. Образовавшееся соединение содержит в молекуле как альдегидную, так и спиртовую группы (отсюда название альд-оль).
2. Конденсация Кляйзена
Ароматические альдегиды взаимодействуют кетонами алифатических углеводородов:
Способы получения
1. Окисление органических соединений (углеводородов, спиртов, альдегидов, кетонов):
2. Гидролиз нитрилов
3. Действие двуокиси углерода на металлорганические соединения:
4. Оксосинтез. Взаимодействие олефинов с оксидом углерода (II) и водяным паром в присутствии катализаторов (тетракарбонил никеля, фосфорной кислоты и др.) при температуре 300–4000С и давлении 2–5∙107 Па дает смесь кислот нормального и изостроения:
5. Гидролиз тригалогенопроизводных, содержащих галоген у одного углеродного атома:
6. Ароматические кислоты могут быть получены окислением гомологов бензола:
7. Кроме того, ароматические карбоновые кислоты могут быть получены:
а) сплавлением ароматических сульфатов с формиатами и цианидами
б) реакцией ароматических соединений с галогенопроизводными угольной кислоты:
Номенклатура и изомерия
Изомерия карбоновых кислот определяется строением радикала, связанного с карбоксильной группой.
По номенклатуре ИЮПАК кислоты называют по соответствующим углеводородам с добавлением окончания –овая и слова кислота, причем нумерация цепи начинается с углерода карбоксильной группы. Однако, в большинстве случаев, используют тривиальные названия.
Иногда при составлении рациональных названий кислот с разветвленной цепью атомов углерода их рассматривают как производные уксусной кислоты, в молекуле которой атомы водорода метильной группы замещены радикалами, например:
Способы получения
1. Окисление органических соединений (углеводородов, спиртов, альдегидов, кетонов):
2. Гидролиз нитрилов
3. Действие двуокиси углерода на металлорганические соединения:

4. Оксосинтез. Взаимодействие олефинов с оксидом углерода (II) и водяным паром в присутствии катализаторов (тетракарбонил никеля, фосфорной кислоты и др.) при температуре 300–4000С и давлении 2–5∙107 Па дает смесь кислот нормального и изостроения:
5. Гидролиз тригалогенопроизводных, содержащих галоген у одного углеродного атома:
6. Ароматические кислоты могут быть получены окислением гомологов бензола:
7. Кроме того, ароматические карбоновые кислоты могут быть получены:
а) сплавлением ароматических сульфатов с формиатами и цианидами
б) реакцией ароматических соединений с галогенопроизводными угольной кислоты:
Физические свойства
В зависимости от строения карбоновые кислоты являются жидкостями или твердыми телами. Уменьшение длины углеродного скелета или появление дополнительной карбоксильной группы увеличивают растворимость кислоты в воде. На физические свойства кислот оказывают влияние ассоциация молекул вследствие образования водородных связей. Кислоты образуют более прочные водородные связи, чем спирты, так как связи О─Н в них в большей степени поляризованы. В твердом, жидком и даже в некоторой степени в парообразном состоянии карбоновые кислоты существуют в виде димеров:
Ароматические кислоты кипят при несколько более высоких и плавятся при значительно более высоких температурах, чем кислоты жирного ряда с тем же числом углеродных атомов. В водных растворах ароматические монокарбоновые кислоты обнаруживают большую степень диссоциации, чем кислоты жирного ряда: константа диссоциации бензойной кислоты 6,6∙10-5, уксусной кислоты 1,75∙10–5.
Химические свойства
Кислотный характер карбоновых кислот ярко выражен. Это объясняется взаимным влиянием атомов в карбоксильной группе: в ней электронная плотность смещена в сторону наиболее электроноакцепторного атома кислорода
Это ослабляет связь между кислородом и водородом и облегчает отделение иона водорода, т.е. диссоциацию кислоты.
Появление пониженной электронной плотности (δ+) на центральном углеродном атоме карбоксила приводит также к стягиванию σ–электронов соседней углерод-углеродной связи к карбоксильной группе и появлению пониженной электронной плотности (δ+) на α–углеродном атоме кислоты.
Сдвиг электронной плотности в молекуле недиссоциированной карбоновой кислоты понижает электронную плотность на гидроксильном атоме кислорода и повышает ее на карбонильном. Этот сдвиг еще больше увеличивается у аниона кислоты.
Результатом сдвига является полное выравнивание зарядов в анионе, названное мезомерией карбоксиланиона. Выравнивание электронной плотности приводит к выигрышу в энергии и для многих реакций является движущей силой, называемой энергией резонанса.
Наличие электрофильных заместителей в радикале, особенно в α–положении, кратных связей, или появление второй карбоксильной группы приводит к возрастанию кислого характера.
1. Карбоновые кислоты способны образовывать соли с металлами, их оксидами, гидроксидами, аммиаком:
2. Характерной реакцией карбоновых кислот является их способность образовывать со спиртами в присутствии минеральных кислот сложные эфиры – реакция этерификации:
Роль катализатора в реакции этерификации играют ионы водорода. Механизм реакции этерификации описывается следующим образом:
а) кислород карбонильной группы кислоты, захватывая протон, образует карбкатион I:
Карбкатион I присоединяет молекулу спирта за счет неподеленных электронных пар атома кислорода с образованием промежуточного тетраэдрического комплекса II, который способен обратимо распадаться с отщеплением воды и образованием нового карбокатиона сложного эфира III. При диссоциации последнего образуется сложный эфир, причем освобождается катализатор протон.
Большой интерес представляет вопрос кислота или спирт отщепляют гидроксил в реакциях этерификации.
С помощью «меченых атомов» (тяжелого изотопа кислорода 18О) было показано, что вода при этерификации образуется за счет водорода спирта и гидроксила кислоты:
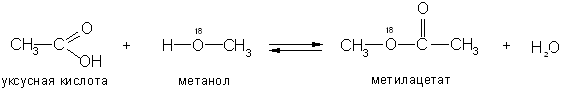
Сложные эфиры получают также взаимодействием галогенацилов со спиртами и алкоголятами и нагреванием солей карбоновых кислот с галогеналкилами и действием спиртов на ангидриды кислот
Сложные эфиры низших кислот и спиртов – жидкости с приятным запахом, в некоторых случаях напоминающих запах плодов. Например: изоамилацетат – грушевая эссенция.
Для сложных эфиров характерны реакции гидролиза и переэтерификации:
3. При действии галогенидов фосфора и серы на карбоновые кислоты образуются галогенангидриды кислот. При этом, как и в случае спиртов, гидроксил замещается галогеном:
Галогенангидриды кислот можно называть по кислоте и галогену, например бромангидрид масляной кислоты. Но чаще их называют по кислотному радикалу - ацилу. Перед названием ацила указывают галоген:
Низшие галогенангидриды – жидкости с весьма резким запахом, раздражающим слизистые оболочки.
При взаимодействии галагенангидридов с соединениями, содержащими атом металла или активный атом водорода, происходит замена его кислотным остатком – ацилом. Подобные реакции называются ацилированием и позволяют получить все производные кислот: ангидриды, кислоты, сложные эфиры, амиды, пероксиды и т.д.
Все эти реакции являются реакциями нуклеофильного замещения и идут в большинстве случаев по тетраэдрическому механизму.
4. При дегидратации кислот или при взаимодействии солей кислот с их галогенангидридами образуются ангидриды кислот:
Ангидриды низших кислот – легкоподвижные жидкости с острым запахом; в воде плохо растворимы или вовсе не растворимы. Кипят при более высокой температуре, чем соответствующие кислоты.
Ангидриды кислот обладают большой химической активностью и являются, как и галогенангидриды, хорошими ацилирующими агентами:
Большое значение имеет реакция получения фторангидрида.
5. При пропускании паров кислот вместе с аммиаком над дегидратирующими катализаторами получаются амиды кислот:
Амиды кислот получают также действием NH3 на галогенангидриды или ангидриды кислот или из аммонийных солей кислот:
Амиды кислот – кристаллические вещества (кроме жидкого амида муравьиной кислоты – формамида).
Амиды, за счет сопряжения свободной электронной пары азота с карбонильным кислородом, почти лишены основных свойств
Наиболее важным для амидов являются реакции:
а) гидролиза в кислой или щелочной среде
б) дегидратации в присутствии пентаоксида фосфора
О нитрилах кислот подробнее смотрите в главе «Азотсодержащие соединения».
6. Галогензамещенные кислоты могут быть получены при действии молекулярного хлора или брома на карбоновые кислоты. Наиболее легко α-галогензамещенные производные кислот образуются из ангидридов и галогенангидридов:
Способы получения
Получение нитросоединений, по М. И. Коновалову, нитрованием в газовой фазе; из галогенопроизводных, а также в ароматическом ряду действием нитрующей смеси рассмотрено в гл.3,8,9
Химические свойства
1. Восстановление. Конечными продуктами восстановления нитросоединений являются первичные амины.
2. Действие щелочей. Таутомерия нитросоединений.
Нитрогруппа, обладая сильным положительным зарядом на атоме азота, оттягивает на себя электронную плотность и увеличивает подвижность водородов у соседнего атома углерода – их способность отщепляться в виде протона. Подвижность α-водородных атомов первичных и вторичных нитросоединений проявляется в их способности реагировать со щелочами с образованием солей. Это объясняется тем, что в щелочной среде нитросоединения перегруппировываются в аци-нитроформу (кислотную):
Таким образом, нитросоединения следует рассматривать как таутомерные вещества, реагирующие в нитро- и аци-нитроформах.
Если щелочные растворы нитросоединений обработать минеральной кислотой, то происходит медленный обратный сдвиг равновесия:
3. Подвижность α-водородных атомов проявляется при взаимодействии первичных и вторичных нитросоединений с альдегидами:
Эта конденсация идет по альдольно-кротоновому типу.
4. Первичные и вторичные нитросоединения реагируют с азотистой кислотой, а третичные не реагируют:
+
Щелочные соли нитроловых кислот в растворе имеют красный цвет. Псевдонитролы в растворах и в расплавах окрашены в синий или зеленовато-синий цвет.
Способы получения
1. Действие аммиака на галогенпроизводные (реакция Гофмана). При этом получается смесь различных аминов:
Смесь аминов обрабатывают щелочью и подвергают перегонке с водяным паром, а гидроксид полностью замещенного аммония [(CH3)4N]+OH− остается в перегонной колбе. Разделение аминов производят, пользуясь их различной реакционной способностью.
2. Пропусканием паров спирта и аммиака при 3000С над катализатором (Al2O3; ThO2) получают смесь первичных, вторичных и третичных аминов с преобладанием первичных:
3. Амиды кислот при расщеплении гипобромитом или гипохлоритом дают первичные амины (перегруппировка Гофмана):
4. Восстановление различных азотосодержащих соединений: нитросоединений, нитрилов, изонитрилов, оксимов или гидразинов:
Химические свойства
В химическом отношении амины очень сходны с аммиаком: вступают в различные реакции как нуклеофильные реагенты. Как и аммиак, амины обладают основными свойствами, что объясняется связыванием протонов в слабо диссоциирующий катион замещенного аммония. Основность ароматических аминов понижена.
1. Амины с минеральными кислотами дают соли, которые под действием более сильного основания вновь дают свободные амины:
2. Амины вступают в реакцию алкилирования.
3. Амины можно ацилировать, в частности ацетилировать, действуя уксусным ангидридом или хлористым ацетилом:
4. Первичные амины могут быть окислены до нитросоединений или других продуктов окисления:
Следовательно, при использовании аминов в различных синтезах, аминогруппу необходимо защищать при действии окислителей. Такой защитой является реакция ацилирования аминов (см