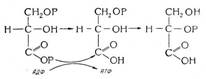
Предмет термодинамики
Предмет термодинамики - изучение законов взаимных превращений различных видов энергии, связанных с переходом энергии между телами в форме теплоты и работы. Химическая термодинамика применяет термодинамические методы для описания химических и физико-химических явлений: химических реакций, фазовых переходов и процессов в растворах.
Различие между теплотой и работой, принимаемое термодинамикой как исходное положение, имеет смысл только для тел, состоящих из множества молекул. Поэтому термодинамика рассматривает лишь макроскопические системы, не принимая во внимание поведение и свойства отдельных молекул.
Объект изучения термодинамики – термодинамическая система, то есть макроскопическая часть пространства, ограниченная реальной или мысленной поверхностью от окружающей среды. По типу взаимодействия системы с окружающей средой различают:
· открытые системы – возможен обмен массой, теплообмен, изменение объема;
· закрытые системы – нет обмена массой, но возможен теплообмен, изменение объема, электрического заряда и т.п.;
· изолированные системы – нет обмена массой, теплотой и нет изменения объема.
Система является гомогенной, если каждый параметр ее имеет во всех частях системы одно и то же значение или непрерывно изменяется от точки к точке; внутри нее нет поверхностей раздела, которые отделяли бы друг от друга части системы, отличающиеся по свойствам. Система является гетерогенной, если она состоит из нескольких макроскопических частей, отделенных одна от другой видимыми поверхностями раздела; на этих поверхностях некоторые параметры изменяются скачком. Однородная система - такая, в которой все участки объема обладают одинаковым составом и свойствами. Неоднородная система может быть и гомогенной, если ее состав и свойства изменяются постепенно, без образования поверхностей раздела.
Фаза - совокупность всех гомогенных частей системы, одинаковых по составу, физическим и химическим свойствам и отграниченных друг от друга поверхностью раздела. Гомогенная система представляет собой одну фазу, гетерогенная содержит не менее двух. Фазы, состоящие из химически индивидуальных веществ - простые (чистые); фазы, состоящие из двух или более веществ - смешанные.
Для описания свойств системы используются специальные термодинамические переменные (или термодинамические параметры). Это физические величины, с помощью которых описывают явления, связанные с взаимными превращениями теплоты и работы. Всё это макроскопические величины, выражающие свойства больших групп молекул.
Состояние любой термодинамической системы может быть охарактеризовано количественно с помощью термодинамических переменных. Все они взаимосвязаны, и для удобства построения математического аппарата их условно делят на независимые переменные и термодинамические функции. Термодинамические функции разделяют на
· функции состояния, которые зависят только от состояния системы и не зависят от пути, по которому это состояние получено;
· функции перехода, значение которых зависит от пути, по которому происходит изменение системы.
Примеры функций состояния: внутренняя энергия U, энтальпия H, энергия Гельмгольца F, энергия Гиббса G, энтропия S. Термодинамические переменные – объем V, давление р, температуру Т – также можно считать функциями состояния, так как они однозначно характеризуют состояние системы. Функции состояния характеризуются следующими свойствами:
1) бесконечно малое изменение функции f является полным дифференциалом и обозначается df;
2) изменение функции при переходе из состояния 1 в состояние 2 определяется только этими состояниями:
 = f 2 – f 1;
= f 2 – f 1;
3) в результате любого циклического процесса функция состояния не изменяется:  = 0.
= 0.
Примеры функций перехода: теплота Q и работа А.
· Состояние системы – совокупность физических и химических свойств, характеризующих эту систему. Изменение каких-либо свойств (даже одного) означает изменение термодинамического состояния системы. Если хотя бы один из параметров системы изменяется, то говорят, что в системе происходит термодинамический процесс.
Энтальпия
Энтальпия Н - это сумма внутренней энергии (U) и внешней (р V):
H = U + р V.
DH = DU + D(р V); при р = const DН = DU + р DV.
Изменение энтальпии в изобарном процессе включает в себя изменение внутренней и внешней энергии, причем изменение внешней энергии равно работе изобарного расширения (сжатия). U - функция состояния; р и V - параметры состояния, их изменение не зависит от пути процесса. Следовательно, энтальпия - также функция состояния, так как ее изменение не зависит от пути процесса.
Если процесс идет при р = const:
dQP = dU + р dV = dU + d (р V) = d (U + р V) = dH.
Тепло, передаваемое системе при р = const, расходуется на приращение энтальпии.
dQP = dH, QP = DH.
Теплота, поглощаемая при р = const, также не зависит от пути процесса.
Если процесс изобарно-изотермический, то DU = 0 и DH = р DV.
C р = 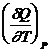 =
=  , dH = C р dT, DH = C р DT.
, dH = C р dT, DH = C р DT.
Введение энтальпии упрощает математическое выражение изобарного процесса. Кроме того, изменение Н может быть во многих случаях легко измерено, вследствие чего эта функция широко применяется при термодинамических исследованиях, особенно изобарных процессов.
Абсолютное значение энтальпии не может быть вычислено с помощью уравнений термодинамики, так как оно включает в себя абсолютную величину внутренней энергии.
2. Энтропия в случае равновесных и неравновесных процессов. Условия равновесия в изолированной системе
Для КПД цикла Карно было получено следующее уравнение:
h =  =
= 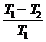 ; 1 -
; 1 - 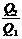 = 1 -
= 1 - 
= или 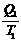 -
- 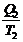 = 0
= 0
Отношение Q/Т - приведенная теплота. Вышеприведенная запись означает: алгебраическая сумма приведенных теплот по обратимому циклу Карно равна нулю.
 Понятие о б/м циклах Карно:
1. Бесконечно мала изотерма, конечна адиабата Понятие о б/м циклах Карно:
1. Бесконечно мала изотерма, конечна адиабата
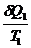 - - 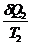 = 0
dQ/Т - элементарная приведенная теплота
2. Бесконечно мала адиабата, конечна изотерма = 0
dQ/Т - элементарная приведенная теплота
2. Бесконечно мала адиабата, конечна изотерма
 = = 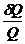 = = 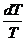
|  P dQ1, T1
Q, T
Q - dQ, T - dT
dQ2, T2
V P dQ1, T1
Q, T
Q - dQ, T - dT
dQ2, T2
V
|
Растворы. Термодинамика многокомпонентных систем, химический потенциал. Уравнения Гиббса – Дюгема. Давление насыщенного пара бинарных жидких растворов. Закон Рауля, идеальные растворы, предельно разбавленные растворы. Отклонения от закона Рауля.
Растворами называются фазы, состав которых можно непрерывно изменять в известных пределах, то есть это фазы переменного состава. Растворы представляют собой однородные смеси молекул (атомов, ионов) двух или более веществ, между которыми имеются физические и химические взаимодействия. Растворы, как правило, термодинамически устойчивы.
Простейшие составные части раствора, которые могут быть выделены в чистом виде и смешением которых можно получить растворы любого состава - компоненты раствора.
Часто деление компонентов на растворитель и растворенное вещество условно. Обычно компонент, находящийся в избытке, называют растворителем, а остальные компоненты - растворенные вещества. Если одним из компонентов раствора является жидкость, а другими - газы или твердые вещества, то растворителем считают жидкость.
Основными параметрами состояния раствора являются р, Т и концентрации - относительные количества компонентов в растворе.
Уравнения Гиббса – Дюгема
dG = Vd р – SdT + Sm i d ni.
При р, T = const dG = Sm i d ni.
Функция G = G(n 1 , n 2 , …) обладает следующим свойством: если массы всех компонентов системы возрастают в одно и то же число раз, то и G возрастает в то же число раз (величина G = U – TS + р V возрастает пропорционально массе, так как U, S, V возрастают пропорционально массе).
Таким образом, интегрируя вышеприведенное уравнение при постоянных соотношениях между массами (постоянный состав раствора), получим:
G = Sm i ni.
При указанных условиях величины m i остаются постоянными в процессе нарастания массы, то есть (при р, Т = const) они зависят только от состава раствора и являются факторами интенсивности. Þ Можно определить химический потенциал как изобарный потенциал системы, приходящийся на 1 моль компонента.
dG = n 1 dm1 + n 2 dm2 + … + ni dm i + m1 d n 1 + m2 d n 2 + … + m i d ni =
= S ni dm i + Sm i d ni.
При р, Т = const S ni dm i = 0. (1)
Для бинарного раствора: n 1 dm1 + n 2 dm2 = 0,
dm2 = –  dm1 = –
dm1 = – 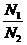 dm1. (2)
dm1. (2)
(N1, N2 – мольные доли компонентов).
Уравнения (1) – (2) – уравнения Гиббса – Дюгема.
Химическое равновесие, общее условие химического равновесия. Закон действующих масс, константа равновесия. Уравнение изотермы химической реакции. Стандартные изобарные потенциалы реакций, их применение. Тепловой закон Нернста, расчет химических равновесий.
Направление химической реакции в ряде случаев зависит от давления газа и от концентрации раствора и при известных значениях этих величин реакция может прекратиться, не дойдя до конца. Т.о., химические реакции обратимы: наряду с химическим взаимодействием между исходными веществами (прямая реакция) протекает химическое взаимодействие между продуктами реакции (обратная реакция), в результате которого снова образуются исходные вещества. По мере протекания процесса скорость прямой реакции (количество молекул, прореагировавших за секунду) уменьшается, а скорость обратной реакции увеличивается. Когда обе скорости сравняются, наступает состояние химического равновесия - число молекул веществ, составляющих химическую систему, перестает меняться и остается постоянным во времени при неизменных внешних условиях. Химическое равновесие является динамичным и подвижным - с изменением внешних условий равновесие сдвигается в одну или в др. сторону; б/м изменение внешних условий влечет за собой б/м изменение состояния равновесия. Т.о., химические реакции могут протекать как термодинамически равновесные процессы, т.е. к ним можно применять общие условия термодинамического равновесия.
Изменение изобарного потенциала G системы, в которой протекает химическая реакция, определяется уравнением:
dG = - SdT + VdP + m1dn1 + m2dn2 +...
Однако в этом случае изменения масс компонентов dn1, dn2... не являются независимыми, а связаны стехиометрическими соотношениями:
n1А1 + n2А2 +... = n1¢А1¢ + n2¢А2¢ +...
Изменения масс компонентов, выраженные в молях, пропорциональны стехиометрическим коэффициентам уравнения реакции, взятым с соответствующим знаком («-» для исчезающих веществ, «+» - для образующихся):
 =
=  =... =
=... = 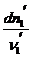 =
=  =... = dc
=... = dc
изменения масс изменения масс
исходных веществ продуктов реакции
Отношение dni /n i одинаково для всех участников химической реакции и может быть записано в форме дифференциала некоторой величины c. c - химическая переменная: показывает массу каждого компонента, вступившую к данному моменту в реакцию и измеренную в эквивалентных единицах, отвечающих уравнению реакции. Химическая переменная характеризует только одну определенную химическую реакцию. Если в системе протекает несколько реакций, то для каждой из них имеется своя химическая переменная (c1, c2...).
dG = VdP - SdT - n1m1dc - n2m2dc -... + n1¢m1¢dc + n2¢m2¢dc +... =
= VdP - SdT + å(n i m i)dc
Здесь G = G (P, T, c),  = ån i m i
= ån i m i
При P,T = const: (¶G)P,T = å(n i m i)dc
Для реакций, протекающих самопроизвольно при P,T = const, dG < 0 Þ å(n i m i) < 0, т.к. dc > 0 (по определению).
Когда реакция находится в состоянии равновесия, функция G = f (c) имеет минимальное значение:
= ån i m i = 0 - это условие химического равновесия (в общей форме)
Аналогично можно вывести:
 = ån i m i = 0 в состоянии равновесия.
= ån i m i = 0 в состоянии равновесия.
Закон действия масс (ЗДМ)
Связь между равновесными концентрациями (или парциальными давлениями) веществ, участвующих в химической реакции, выражается законом действия масс, количественная формулировка и кинетический вывод которого были даны Гульдбергом и Вааге (1867).
n1А1 + n2А2 Û n1¢А1¢ + n2¢А2¢
v 1 = k 1  - скорость прямой реакции
- скорость прямой реакции
v 2 = k 2  - скорость обратной реакции
- скорость обратной реакции
v 1 = v 2 в состоянии равновесия
k 1 = k 2
КС = k 1 / k 2 = /
(КС - константа равновесия, выраженная через концентрации)
ЗДМ можно вывести из уравнения ån i m i = 0, если химические потенциалы выразить как функции концентраций, парциальных давлений и т.д. компонентов, участвующих в реакции:
m i = Gi (T) + RT ln C i 
m i = Gi ¢(T) + RT ln P i - если компоненты - идеальные газы
m i = Gi ¢¢(T,P) + RT ln N i
m i = m i (T) + RT ln fi - если компоненты - реальные газы
m i = m i o (T) + RT ln N i - если компоненты - идеальные растворы
m i = m i o (T) + RT ln ai - если компоненты - реальные растворы
Выведем ЗДМ для газовой реакции, если компоненты - идеальные газы. Исходное уравнение m i = Gi ¢(T) + RT ln P i подставим в уравнение ån i m i = 0.
ån i Gi ¢(T) + RT ån i ln р i , равн = 0
ån i ln р i , равн = -  = f (T)
= f (T)
Опустим индекс (равн); заменим сумму логарифмов логарифмом произведения р i; а f (T) - логарифмом некоторой функции КР (Т):
ln  = - = ln KP (T)
= - = ln KP (T)
= 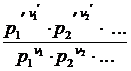 = KP (T)
= KP (T)
Величина КР, выраженная через равновесные парциальные давления в идеальной газовой смеси, есть функция только Т и не зависит от суммарного Р и парциальных давлений компонентов в исходной смеси. При T = const KP = const.
КР - константа химического равновесия, а уравнение называется законом действия масс.
Если газ - реальный, то таким же путем получим:
K f =  ; K f ® KP при Р ® 0
; K f ® KP при Р ® 0
Необходимо различать константы равновесия, выраженные разными способами, т.к. их числовые значения неодинаковы. К выражают: через р i, с i, N i.
KP =  ; KC =
; KC =  ; KN =
; KN = 
Связь между ними можно установить, используя уравнения для идеальной газовой смеси: P i = C i RT Þ KP = KC (RT)Dn
P i = N i P Þ KP = KN PDn
Dn = n1¢ + n2¢ + n3¢ +... - n1 - n2 - n3 -...
Т.к. КР не зависит от Р (для идеальных газов), то и КС от него не зависит. KN же зависит от Р и не зависит от исходных количеств компонентов.
Если Dn = 0, т.е. реакция протекает без изменения числа молекул, то КР = КС = КN.
Основной постулат химической кинетики. Скорость химической реакции, скорость реакции средняя и истинная. Кинетическая классификация реакций, различие понятий «порядок реакции» и «молекулярность реакции», понятие об элементарной реакции. Необратимые реакции первого, второго, n-го и нулевого порядка.
В общем виде химическую реакцию можно записать следующим образом:
n1 А1 + n2 А2 +... + nn An = n1¢A1¢ + n2¢A2¢ +... + nn¢An¢.
Скоростью химической реакции называется количество молекул данного сорта, реагирующих в единицу времени в единице объёма. Для определения скорости химической реакции достаточно знать изменение во времени количества одного из участвующих в реакции веществ (исходного или конечного), так как изменение количества всех остальных веществ можно определить из уравнения реакции.
Химические реакции редко протекают в одну стадию, так, как их принято записывать. Запись уравнения реакции можно рассматривать как символическое выражение материального баланса (закона сохранения вещества). В действительности подавляющая часть химических реакций протекает через ряд промежуточных стадий, и в большинстве случаев детальный механизм протекания реакции нам неизвестен из-за больших трудностей выявления промежуточных продуктов. Скорость суммарной реакции определяется скоростью наиболее медленной стадии, которая называется лимитирующей.
Еще в самом начале исследований по химической кинетике было сделано физически очевидное предположение, что реагируют только те молекулы, которые сталкиваются. Как известно, число столкновений прямо пропорционально числу молекул, поэтому скорость реакции должна быть пропорциональна концентрации реагирующих веществ. В общем случае можно принять, что скорость химической реакции прямо пропорциональна концентрациям реагирующих веществ в некоторых степенях:
v = k  , [ v ] = моль/(м3×с) или молекул/(м3×с);
, [ v ] = моль/(м3×с) или молекул/(м3×с);
k - константа скорости химической реакции, k зависит от природы реакции и является функцией температуры;
ni - порядок реакции по данному веществу; в общем случае порядки реакции по веществам никак не связаны со стехиометрическими коэффициентами n i этих веществ в уравнении реакции, и только в тех случаях, когда реакция протекает в одну стадию, порядок реакции ni по данному веществу совпадает со стехиометрическим коэффициентом n i данного вещества в химическом уравнении.
Вышеприведенное выражение называют основным постулатом химической кинетики.
Физический смысл коэффициента k: v = k, если 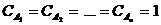 , то есть константа скорости химической реакции есть скорость реакции при условии, что концентрации реагирующих веществ равны 1; k иногда называют удельной скоростью химической реакции. Размерность константы скорости зависит от порядка реакции.
, то есть константа скорости химической реакции есть скорость реакции при условии, что концентрации реагирующих веществ равны 1; k иногда называют удельной скоростью химической реакции. Размерность константы скорости зависит от порядка реакции.
Скорость реакции является функцией времени, так как количества реагирующих веществ меняются во времени. Введем понятие средней скорости реакции:
 ,
,
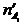 и
и  - число молей одного из веществ в начальный t¢ и конечный t¢¢ моменты времени; V - объем системы. Знак «-» в выражении стоит тогда, когда мы следим за ходом реакции по изменению количества одного из исходных веществ, поскольку в этом случае > , а скорость реакции может быть только положительной величиной. Знак «+» используют, если скорость реакции определяют по ее продукту.
- число молей одного из веществ в начальный t¢ и конечный t¢¢ моменты времени; V - объем системы. Знак «-» в выражении стоит тогда, когда мы следим за ходом реакции по изменению количества одного из исходных веществ, поскольку в этом случае > , а скорость реакции может быть только положительной величиной. Знак «+» используют, если скорость реакции определяют по ее продукту.
Если t¢¢ - t¢ ® 0, то получим истинную скорость v реакции (скорость реакции в данный момент времени):
v = ±  .
.
Если V = const, то v = ±  .
.
Этим уравнением удобно пользоваться при рассмотрении реакций в растворах, так как изменением объема раствора в результате реакции во многих случаях можно пренебречь.
Скорость реакции зависит от природы реагирующих веществ, их концентрации, температуры и наличия катализатора.
Следует иметь в виду, что при вышеприведенном определении скорости химической реакции (число молекул, реагирующих в единицу времени в единице объема) числовое значение величины скорости реакции будет зависеть от того, какое вещество из участвующих в реакции мы выбрали, чтобы следить за ее скоростью, так как стехиометрические коэффициенты разных участников реакции могут отличаться друг от друга. Например, в реакции синтеза бромоводорода
Н2 + Br2 = 2HBr
за одно и то же время образуется в 2 раза большее число молей HBr, чем прореагирует молей Н2 и Br2 , и, следовательно, скорость реакции, определенная по бромоводороду, будет в 2 раза выше, чем скорость, определенная по водороду либо по брому:
 .
.
Чтобы устранить эту неопределенность, скорость реакции рассчитывают на единицу стехиометрического коэффициента, и тогда ее величина не зависит от того, по какому веществу ее определяют:
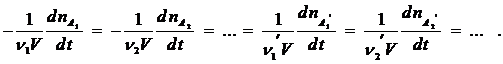
В общем виде можно записать:
 где
где 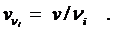
Реакции нулевого порядка
Такой порядок получается при постоянной скорости реакции, что возможно при поддержании постоянной концентрации исходных веществ. Нулевой порядок встречается главным образом в гетерогенных и фотохимических реакциях.
 = k, dx = k dt, x = kt + const.
= k, dx = k dt, x = kt + const.
При t = 0 x = 0 Þ x = kt.
Удельная и эквивалентная электропроводность, ее зависимость от концентрации и температуры. Подвижность ионов, закон Кольрауша, формула Стокса. Аномальная подвижность ионов гидроксония и гидроксила (механизм).
Электропроводность К - величина, обратная электрическому сопротивлению R. Так как R = r  , то К =
, то К =  ×
×  = k
= k
где r - удельное электрическое сопротивление; l - расстояние между электродами; S - площадь электрода; k - удельная электропроводность.
Удельная электропроводность k жидкости - это электропроводность одного кубического сантиметра раствора, заполняющего пространство между плоскими электродами одинаковой, очень большой площади, находящимися на расстоянии 1 см. Кубический сантиметр раствора должен находиться вдали от границ электрода. [k] = Ом–1× см–1.
Кривая зависимости удельной электропроводности растворов от концентрации обычно имеет максимум (четко выраженный для сильных электролитов и сглаженный для слабых). Наличие максимумов на кривых k - с можно объяснить следующим образом. Электропроводность растет пропорционально числу ионов, которое, в свою очередь, растет с концентрацией, но существуют и факторы противоположного действия. В концентрированных растворах сильных электролитов ионная атмосфера существенно уменьшает скорость движения ионов, и электропроводность падает. В слабых электролитах плотность ионной атмосферы мала и скорость движения ионов мало зависит от концентрации, однако с увеличением концентрации раствора заметно уменьшается степень диссоциации, что приводит к уменьшению концентрации ионов и падению электропроводности.
 k сильный эл-т
слабый эл-т
С
Зависимость удельной электропроводности от концентрации электролита k сильный эл-т
слабый эл-т
С
Зависимость удельной электропроводности от концентрации электролита
|  l
сильный эл-т
слабый эл-т
С
Зависимость эквивалентной электропроводности от концентрации электролита
l
сильный эл-т
слабый эл-т
С
Зависимость эквивалентной электропроводности от концентрации электролита
|
Удельная электропроводность зависит от температуры. Зависимость дается эмпирическим уравнением: kt = k18 × [1 + a (t - 18)]
a - температурный коэффициент электропроводности (a > 0); k18 (k25) - стандартное значение. Коэффициент a зависит от природы электролита. В случае слабых электролитов a больше, чем для сильных. Следует отметить, что температурные коэффициенты электропроводности водных растворов и вязкости воды близки по своей величине, но обратны по знаку. Это свидетельствует о том, что увеличение удельной электропроводности с ростом температуры связано, главным образом, с уменьшением вязкости раствора.
Эквивалентная электропроводность l [в см2/(г-экв×Ом)] - это электропроводность такого объема (j см3) раствора, в котором содержится 1 г-экв растворенного вещества; раствор заполняет пространство между плоскими электродами одинаковой, очень большой площади, находящимися на расстоянии 1 см.
Связь между k и l дается уравнениями
l = k× j; l = 
где величина j, равная 1000/с см3/г-экв, называется разведением.
Мольная электропроводность электролита m - это произведение эквивалентной электропроводности на число грамм-эквивалентов в 1 моль диссоциирующего вещества.
 l
сильный эл-т
слабый эл-т l
сильный эл-т
слабый эл-т

|
 l сильный эл-т
l¥
слабый эл-т
j l сильный эл-т
l¥
слабый эл-т
j
|
Зависимость эквивалентной электропроводности от концентрации:
1. Зависимость l - с: с увеличением с величина l уменьшается сначала резко, а затем более плавно.
2. Зависимость l -  : для сильных электролитов в области малых концентраций соблюдается медленное линейное уменьшение l с увеличением , что соответствует эмпирической формуле Кольрауша (закону квадратного корня):
: для сильных электролитов в области малых концентраций соблюдается медленное линейное уменьшение l с увеличением , что соответствует эмпирической формуле Кольрауша (закону квадратного корня):
l = l¥ - А
l¥ - предельная эквивалентная электропроводность при бесконечном разведении: с ® 0, j ® ¥. А – эмпирическая постоянная. При несколько более высоких концентрациях сильных электролитов лучшее согласие с опытом дает уравнение, известное под названием закона кубического корня:
l = l¥ - А 
Для разбавленных растворов слабых электролитов вышеприведенные законы не соблюдаются.
3. Зависимость l - j: значение l сильных электролитов растет с увеличением j и асимптотически приближается к l¥. Для слабых электролитов значение l также растет с увеличением j, но приближение к пределу и величину предела в большинстве случаев практически нельзя установить.
Подвижность ионов
Электропроводность электролитов зависит от следующих факторов: в первую очередь, от природы растворителя (вязкость, диэлектрическая проницаемость) и природы растворенного вещества (размера и заряда ионов), затем от напряженности поля, концентрации электролита, температуры и некоторых других. Для количественной характеристики влияния природы иона на величину электропроводности введены следующие понятия: абсолютная подвижность ионов, подвижность ионов (ионная электропроводность), предельная подвижность ионов.
Итак, скорости движения катионов u¢ (см/с) и анионов v¢ (см/с) зависят от природы ионов, напряженности электрического поля U/ l, концентрации, Т, вязкости среды и т.п. Пусть все факторы постоянны, кроме напряженности электрического поля; можно считать, что скорость ионов пропорциональна приложенной силе – напряженности поля:
u¢ = u  , v¢ = v
, v¢ = v
u, v - скорости ионов в стандартных условиях, т.е. при напряженности поля, равной 1 В/см; они называются абсолютными подвижностями ионов и измеряются в см2/(с×В).
u×F и v×F - это скорости движения ионов, выраженные в электростатических единицах; они называются ионными электропроводностями (или просто подвижностями ионов):
u×F = l+, v×F = l–
Для сильных электролитов: l = l+ + l–
Для слабых электролитов: с+ = с×a, с– = с×a, l = (l+ + l–)×a
При бесконечном разведении (j ® ¥, a ® 1, с+ = с– = с):
l¥ = lо+ + lо–
- как для сильных, так и для слабых электролитов. Величины lо+ и lо– являются предельными электропроводностями (предельными подвижностями) ионов. Они равны эквивалентным электропроводностям катиона и аниона при бесконечном разведении и измеряются в тех же единицах, что и l и l¥, т.е. в см2/(Ом×г-экв). Вышеприведенное уравнение является выражением закона Кольрауша: эквивалентная электропроводность при бесконечном разведении равна сумме предельных подвижностей ионов.
Т.о., для всех электролитов можно записать:
lс = aс×l¥, aс = lс / l¥
l+ и l– зависят от концентрации (разведения), особенно для сильных электролитов; lо+ и lо– - табличные величины. Все эти величины относятся к 1 г-экв ионов.
Подвижность является важнейшей характеристикой ионов, отражающей их специфическое участие в электропроводности электролита. В водных растворах все ионы, за исключением ионов Н3О+ и ОН–, обладают подвижностями одного порядка; их lо составляют не более 80 см2/(Ом×г-экв) при 25оС; их абсолютные подвижности (u и v) равны нескольким см в час. Подвижности же ионов Н3О+ и ОН– составляют соответственно ~350 и ~200 см2/(Ом×г-экв).
Рассмотрим влияние природы иона (его радиуса и заряда) на величину подвижности иона. Движение иона можно уподобить движению макроскопического шарика в вязкой среде и применить в этом случае формулу Стокса:
u¢ = 
где е - заряд электрона; z - число элементарных зарядов иона; r - эффективный радиус иона (радиус гидратированного иона!), h - коэффициент вязкости; U/ l - напряженность поля.
Движущую силу - напряженность поля U/ l при вычислении абсолютных подвижностей принимаем равной единице. Следовательно, скорость движения ионов обратно пропорциональна их радиусу. Рассмотрим ряд Li+, Na+, K+. Так как в указанном ряду истинные радиусы ионов увеличиваются, то подвижности должны уменьшаться в той же последовательности. Однако в действительности это не так. Подвижности увеличиваются при переходе от Li+ к K+ почти в два раза. Из этого можно сделать заключение, что в растворе и ионной решётке ионы обладают разными радиусами. При этом чем меньше истинный (кристаллохимический) радиус иона, тем больше его эффективный радиус в электролите. Это явление можно объяснить тем, что в растворе ионы не свободны, а гидратированы. Тогда эффективный радиус движущегося в электрическом поле иона будет определяться в основном степенью его гидратации, то есть количеством связанных с ионом молекул воды.
Связь иона с молекулами растворителя ионно-дипольная, а так как напряжённость поля на поверхности иона лития гораздо больше, чем на поверхности иона калия, то степень гидратации иона лития больше степени гидратации иона калия, и эффективный радиус иона лития будет больше, чем у иона калия. Согласно формуле Стокса многозарядные ионы должны обладать большей подвижностью, чем однозарядные. Однако скорости движения многозарядных ионов ненамного превышают скорости движения однозарядных, что объясняется большей степенью их гидратации.
Гальванические элементы. ЭДС. Связь ЭДС с константой равновесия реакции. Электродный потенциал. Диффузионный потенциал. Термодинамический вывод формулы Нернста для электродного потенциала. Стандартный электродный потенциал.
При прохождении электрического тока через электролит на поверхности электродов протекают электрохимические реакции. Протекание электрохимических реакций может порождаться внешним источником тока. Возможно и обратное явление: электрохимические реакции, протекающие на двух электродах, опущенных в электролит, порождают электрический ток, причем реакции идут только при замкнутой цепи (при прохождении тока).
Электрохимическим (или гальваническим) элементом называется устройство для получения электрического тока за счет электрохимических реакций. Простейший электрохимический элемент состоит из двух металлических электродов (проводников первого рода), опущенных в электролит (проводник второго рода) и соединенных между собой металлическим контактом. Несколько электрохимических элементов, соединенных последовательно, образуют электрохимическую цепь.
Важнейшей количественной характеристикой электрохимического элемента является электродвижущая сила (ЭДС, Е), которая равна разности потенциалов правильно разомкнутого элемента (такого, у которого к конечным электродам элемента присоединены проводники первого рода из одного и того же материала).
Если при прохождении электрического тока в разных направлениях на по