Синдром Дауна

 Этиология
Этиология
Собственные причины не установлены. Есть разные теории, но они объясняют не все случаи. В любом случае это мутация.
Кариотип:
· 95% - трисомия по 21 хромосоме
· 4-5% - трансклокации (перенос участка хромосомы на негомологичную хромосому).
У 21 хромосомы на плече есть критический локус, который приводит к Синдрому Дауна.
96 – 98 % – полный вариант транслокации или трисомии, остальные — мозаичные варианты. При мозаичных вариантах клиническая картина более мягкая.
Эпидемиология
Частота — 1 случай на 650-1000 новорожденных; общая частота — 1 случай на 4000 новорожденных.
Синдром Дауна составляет примерно 10% от всех случаев клинически выраженной УО. Риск развития плода и рождении ребёнка синдромом очень зависит от возраста родителей (наиболее важен возраст матери).
Риск рождения ребенка с синдромом Дауна у матери:
· 25 лет — 1:2000 родов;
· 35 лет — 1:385 родов;
· 40 лет — 1:105 родов;
· старше 40 лет — 1:30 родов.
При этом большинство детей с синдромом Дауна рождаются у молодых мам.
Практически все трисомные варианты не наследуются от родителей, а транслокации: 75% приводят к новым мутациям, а 25% - наследуются.
Клиническая картина
Соматические аномалии: низкий рост, укороченные конечности, неправильная поза, своеобразная походка, расстояние между большим и вторымпальцем на ноге, складки на ладонях, небольшая микроефалия, уплощенный затылок, брахицефалия (череп вверх вытянуть больше, чем стороны), уплощенное лицо, небольшие ушные раковины несколько аномальной формы, специфическое лицо, монголоидный разрез глаз, эпиканты (складки у внутренних уголков глаз), аномалии развития радужки, макроглассия (увеличенный язык), растянута передяя брюшная стенка.
Внутренние органы: пороки развития сердца, кишечника, нарушение обмена веществ, умеренная гипофункция щитовидной железы, склонны к полноте, снижен иммунитет, повышенная склонность к злокачественным новообразованиям и лейкозам, подавляющее большинство стерильны (бесплодны).
НПР: а номалии развития НС (в частности ГМ), меньше толщина коры, нейронов и их связей, кондуктивная тугоухость, мышечная гипотания (пониженное давление), косоглазие, нарушение артикуляции, УО, обидчивы, послушны, эмпатийны, хорошие исполнители, друзья. Наблюдается истинная прогредиентность дефекта
Продолжительность жизни: 30 – 40 лет, могут до 50 лет. 25% родившихся погибают на первом году жизни. Основная причина гибели: декомпенсация пороков развития, лейкоз, ускоренное старение.
27. Синдром Тернера (синдром Шерешевского-Тернера)
 Этиология неизвестна. Наследственность.
Этиология неизвестна. Наследственность.
Кариотип характеризуется отсутствием 2 половой хромосомы. Возможны варианты. Во-первых, может наблюдаться мозаицизм (X0, X-моносомия). Во-вторых, вместо кариотипа 45X0 происходят аномалии одной X-хромосомы (структурные аномалии: делеции, кольцевые аномалии и др.). В кариотипе одна X-хромосома либо отсутствует, либо неполноценна (по Y-хромосоме такого нет), поэтому синдром наблюдается ТОЛЬКО У ДЕВОЧЕК, т. к. X-хромосома жизненно необходимо, а Y-хромосома маленькая, где мало генов, и если мутация происходит по Y при этом синдром (моносомия по гоносомам), то это приводит к смерти зародыша. С X при этом синдроме происходит следующее. В клетке активна только одна X-хромосома.
Эпидемиология
Частота — 1 случай на 2000-5000 девочек. Примерно такая же частота в женской популяции.
Вероятность обнаружить этот синдром выше, если обследовать группу женщин с первичной аменореей (отсутствие менструаций).
Клиническая картина
Соматические аномалии: короткая широкая шея с крыловидными складками, низкий рост волос на затылке, антимонголоидный разрез глаз, эпиканты, гипертелоризм (ненормальное увеличенное расстояние между 2 парными органами), аномалии зубов, профиль Сфинкса, широкая грудная клетка, широки и низко расположены соски, низкие ушные раковины, низкий рост (ниже 140 см), лимфатические отеки нижних конечностей, разнообразные доброкачественные дисплазии (избыточность чего-либо; в основном кожи).
Внутренние органы: половой инфантилизм, не развиваются вторичные половые органы, нет менструаций, большинство стерильны, дефекты слуха, зрения, пониженный мышечный тонус.
НПР: диапазон интеллектуального уровня широкий (у значительной части больных УО не формируется, интеллект в низкой норме или ПИН; у части — УО, как правило, легкая, а тяжелая уже при неблагоприятных условиях, если синдром еще чем-то осложнен), зрительно-пространственные нарушения (нарушение математических функций), психический инфантилизм, эмоции развиты (преобладает аффективный фон), трудолюбивы, послушны, высоко адаптированы, на 2 десятилетии жизни становятся критичны к своему дефекту, но из-за эмоциональных особенностей не сильно за это переживают.
Продолжительность жизни существенно не сокращена.
Синдром Клайнфельтера
 Этиология
Этиология
Х-полисомия у мужчин.
Кариотип
Особи мужского пола имеют дополнительную Х-хромосому. Обычно женщины имеют пару ХХ хромосом, а мужчины пару ХY хромосом, однако при этом заболевании мужчины имеют по крайней мере две Х-хромосомы и хотя бы одну Y хромосому.
Эпидемиология
Частота — 1:500 новорожденных мальчиков.
Диагностируется поздно, т. к. проявляется только после полового созревания.
Клиническая картина
Соматические аномалии: рост выше среднего, узкая грудная клетка, узкий лоб, вторичные половые признаки либо не формируется, либо формируется по женскому типу.
Внутренние органы: врожденные пороки сердца, склонность к диабету, полноте, остеопороз, склонность к возникновению злокачественных образований (например молочных желез).
НПР: легкая УО, испытывают трудности в выражении своих мыслей, эмоционально-волевые нарушения, повышенная внушаемость, назойливость, повышенный риск алкоголизации, гомосексуализма.
Продолжительность жизни может быть сокращена из-за проблем со здоровьем, которые могут привести к ранней смерти.
Синдром кошачьего крика
 Этиология и кариотип
Этиология и кариотип
Отсутствие короткого плеча 5 хромосомы.
Эпидемиология
Частота — 1:45000 новорожденных.
Клиническая картина
Соматические аномалии: округлые лицо в детстве (с возрастом черты грубеют), эпиканты, низкие уши, микрогнатия (недоразвитие нижней челюсти), низкая масса тела, общая отставание в развитии, косолапость, микроцефалия, короткая шея, антимонголоидный разрез глаз, косоглазие, глазной гипертелоризм, аномалии развития пальцев (синдактилия (сращение пальцев)), клинодактилия (искривление пальцев в суставах)).
Внутренние органы: аномалии развития (вследствие смерть наступает в первые 15 лет жизни), аномалии гортани (из-за этого плач детей становится похожим на кошачий), пониженный мышечный тонус, грыжи.
НПР: возможно формирование УО,эмоциональная лабильность.
Продолжительность жизни значительно укорочена. Некоторые погибают в первый год жизни.
Синдромы Эдвардса и Патау
Синдром Эдвардса

Этиология
Возникает спорадически. Влияют различные факторы (от окружающей среди до наследственности).
Кариотип
Трисомия по 18 хромосоме.
 Эпидемиология
Эпидемиология
Частота — 1:3000 зачатий; 1:6000 новорожденных. Риск увеличивается в возрастом. У девочек встречается значительно чаще, чем у мальчиков, что связано, возможно, с большей жизнестойкостью женского организма.
Клиническая картина
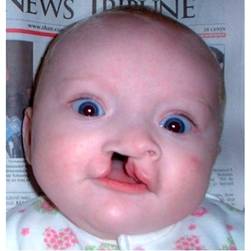
Соматические аномалии: микроцефалия, лоб узкий, широкая переносица, нарушения зрения, выступающий и широкий затылок, низкий вес (2200 г среднем), аномалий мозгового и лицевого черепа, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие, ушные раковины деформированы, располагаются низко, мочек нет, грудь короткая и широкая, аномалии развития стопы (пятка резко выступает, большой палец толстый и короткий),борозды на ладонях, косоглазие, расщелены губы.
Внутренние органы: суставы нормально не функционируют, склонность к вывихам, порок сердца, нарушения обменных процессов, пониженный мышечный тонус, аномальное развитие кишечника, почек, патологии развития половых органов.
НПР: УО, сглаживание или атрофия мозговых извилин, недоразвитие мозжечка и мозолистого тела, задержка НПР.
Продолжительность жизни
Большинство детей не доживают до года, до этого возраста доживают лишь 10% детей. Примерно 50% больных умирают уже в течение первых 2 месяцев, при этом наблюдается связь с половой принадлежностью. Мальчики с этим синдромом живут около 60 суток, а девочки – примерно 280 дней.
 Синдром Патау
Синдром Патау
Этиология
Возникает спорадически. Влияют различные факторы (от окружающей среди до наследственности).
Кариотип
Трисомия по 13 хромосоме.
Эпидемиология
Частота — 1:7000-10000 новорожденных.
Клиническая картина
Соматические аномалии: низкий вес (2500 г), умеренное микроцефалия, низкий, плоская, запавшая переносица, скошенный лоб, суженные глазные щели, расщелины верхней губы и неба, полидактилия, синдактилия, короткая шея, низкие и деформированные уши, аномалии развития половых органов
Внутренние органы: гидроцефалия, грыжи, пороки сердца, пищеварительной системы, кисты поджелудочной.
НПР: ЗПР, аномалии развития ГМ, УО.
Продолжительность жизни
95% больных умирает на первом году жизни. В развитых странах количество детей, доживающих до 5 лет, не превышает 15%, до 10 лет – 2-3%.
Синдром X-трисомии
Этиология
Данный синдром возникает при наличии добавочной Х-хромосомы. Это связано с нарушением расхождения хромосом на ранних стадиях формирования клеток.
Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор мало изучены.
Кариотип
Заболевание, характерное для женщин, в клетках у которых содержится дополнительная половая Х-хромосома.
Эпидемиология
Частота — 1:1000 новорожденных девочек.
Клиническая картина
Соматические нарушения: могут внешне выглядят здоровыми, а может развиваться отставание в росте, деформация костей, уменьшение размеров головы, неправильный рост зубов, патологии зрения.
Внутренние органы: вторичнаяаменорея (нерегулярные менструации с болью), способны к деторождению.
НПР: в большинстве случаев интеллект сохранен, но может развиваться легкая УО, склонность к шизофрении и эпилепсии.
Продолжительность жизни не сокращена.
32. Синдром XYY
Этиология
Нерасхождение хромосом во втором делении отцовского мейоза, с образованием сперматозоида YY. Причины нерасхождения неизвестны.
Кариотип
Наличие дополнительной Y-хромосомы у мужчин.
Эпидемиология
Частота — 1:1000 новорожденных мальчиков.
Клиническая картина
Соматические нарушения: внешне ничем не отличаются от нормы, быстро растут в детстве, могут страдать нарушением координации движений.
Внутренние органы: нет нарушений, могут иметь детей.
НПР: нарушения поведения (гиперактивность, импульсивны, эмоционально незрелые), интеллект в пределах нормы, но ниже, чем у нормальных братьев/сестер, имеют трудности при обучении, особенно письму и чтению
Продолжительность жизни не сокращена.
 33. Синдром Нунан
33. Синдром Нунан
Этиология
Возникает спорадически, но описаны и семейные случаи. Наследуется по аутосомно-доминантному типу. Похож на синдром Тернера. Второе название: псевдо Тернер, синдром Тернер-фенотип с нормальным кариотипом.
Кариотип
Ген локазилован на 12 хромосоме.
Эпидемиология
Частота — 1:1000-2500 новорожденных. Распределение по полу примерно одинаковое.
Клиническая картина
Соматические нарушения: низкий рост, антимонголоидный разрез глаз, эпиканты, гипертелоризм, низко расположенные уши, крыловидные складки на шее. Признаки как у Тернера.
Внутренние органы: геморрагический диатез, повышенная склонность к кровотечениям, аномалии развития сердца и сосудов, репродуктивное развитие может быть в норме, а может у женщин развиваться первичная аменорея, а у мужчин — отсутствовать шустрые сперматозоиды, в этом случае больные стерильны.
НПР: часто нарушение слуха, нетяжелые моторные нарушения, интеллектуальный уровень приходится на широкий диапазон (от УО до ПИН или низкой нормы), значительные нарушения поведения, активности и внимания, речь хорошо развита.
Продолжительность жизни существенно не сокращена.
 34. Синдром фрагильной X-хромосомы (синдром Мартина-Белл)
34. Синдром фрагильной X-хромосомы (синдром Мартина-Белл)
Этиология и кариотип
Наследуется как X-сцепленный доминантный признак. На длинном плече X-хромосомы расположен участок, в котором могут накапливаться нуклеотидные повторы. Если повторов менее 55, то это не патология. Если их от 55-200 — премутация (либо не проявляется, либо проявляется очень легко). Если их больше 200 — мутация. Добавление лишних нуклеотидов может происходит только в женском организме в процессе овогенеза. Мужчина же может передать хромосому с премутацией, близкой к критической, только здоровым сыновьям. У женщин мутация выйдет из состояниями премутации. Если она передаст это хромосому сыну, то у него будет отмечаться этот синдром. Если дочери, то она либо будет здорова, либо у нее будет наблюдаться легкая картина данного синдрома. Поэтому синдром отмечается чаще у мальчиков.
Эпидемиология
Частота — 1:6000 новорожденных девочек; 1:4000 новорожденных мальчиков.
Клиническая картина У МАЛЬЧИКОВ
Соматические нарушения: высокий рост, с возрастом склонность к полноте, большие конечности, крупные гиперпластичные суставы, в пубертатном периоде отмечается макроорхизм (большие яички), гиперпластичная кожа, высокий лоб, вытянутое лицо, массивная нижняя челюсть, выраженные подбородок, большие и оттопыренные уши, большое рот, с возрастом черты лица грубеют.
Внутренние органы: пороки сердца.
НПР: макроэнцефалия (большой ГМ), приводящая к макроцефалии (большая голова), не происходит редуцирование межнейронных связей, ГМ оказывается перегружен, УО (разные степени), гиперкинез (непроизвольные движения различных групп мышц), нарушение внимания и активности, энурез (недержание мочи), энкопрез (недержание кала), дизартрия, заикание,ускоренная речь с персеверацией в виде повторения коротких фраз или их окончаний, агрессивное поведение, 10-15% - эпилепсия, приводящая к атипичному аутизму.
Клиническая картина У ДЕВОЧЕК (проявляется не всегда)
Соматические нарушения: нет
Внутренние органы: нет
НПР: невысокий интеллект (IQ=50-110)
Продолжительность жизни У ДЕВОЧЕК существенно не изменена, У МАЛЬЧИКОВ — ниже.
35. Синдром Вильямса (синдром эльфийского лица)


Этиология и кариотип
Вызван генетической мутацией, приводящей к потере сегмента плеча 7 хромосомы. Что провоцирует делецию хромосомы и является причиной синдрома Вильямса в настоящее время точно неизвестно. Родители больного далеко не всегда обладают семейной историей заболевания. Хромосомное нарушение спорадически. Но в случае констатации заболевания у кого-то из родителей вероятность передачи синдрома ребенку составляет около 50%. При зачатии ребенку с половыми клетками матери или отца передается нарушенная хромосома из 7 пары, лишенная небольшого участка, его недостача является причиной главных особенностей синдрома Вильямса.
Эпидемиология
Частота — 1:10000-20000новорожденных
Клиническая картина
Соматические нарушения: «лицо эльфа», большой рот с полными губами, широкий лоб, короткий нос с тупым и широким кончиком, плоская переносица, ярко-голубые глаза с голубоватыми белками, отечные веки, полные щеки, опущенные к низу, небольшой острый подбородок, нарушен прикус, худые, отставание в росте, плоскостопия, косолапость, узкая грудная клетка, низкая талия, удлиненная шея, склоннсть к ожирению с возрастом, нарушения зрения.
Внутренние органы: сердечно-сосудистые заболевания, поражение желудочно-кишечного тракта, приводящее к урологии и проблемам с суставами.
НПР: нарушение равновесия и координации движений, УО (разные степени), отсутствие способности к наглядно-образному мышлению, концентрированию, планированию, организации деятельности, с трудом овладевают письмом и счетом, имеют развитую речь, добродушны, смешливы, коммуникабельны, инфантильны, неадекватны в поведении, свойственны фобии.
Продолжительность жизни незначительно снижена.
 36. Синдром Рубинштейна-Тейби
36. Синдром Рубинштейна-Тейби
Этиология
Аутосомно-доминантное заболевание. Причины неизвестны. Могут возникать спорадически, но также описаны семейные случаи.
Кариотип
Изменения в 16 хромосоме.
Эпидемиология
Частота — 1:25000 новорожденных.
Клиническая картина
Соматические нарушения: аномалии развития пальцев, микро- или брахицефалия (короткоголовость), расширение переносицы, эпиканты, клювовидный нос, высокое арковидное нёбо, изменение формы и положения ушей, гирсутизм (рост волос у женщин по мужскому типу на лице и теле), низкий рост, искривление позвоночника, деформация грудной клетки, на лице гримаса улыбки.
Внутренние органы: патологии легких, сердца, почек и мочевыделительных путей, пищеварительной системы, риск возникновения онкологии
НПР: УО, затрудненное развитие речи, моторных навыков, нарушение внимания, резкие перепады настроения, хорошо идут на контакт, социализируются, нарушение координации движений, приступы судороги,
Продолжительность жизни зависит от состояния здоровья. Возможна смерть и в раннем возрасте.

 37. Синдром де Ланге
37. Синдром де Ланге
Этиология и кариотип
Причины и развитие болезни до сих пор не известны. Есть предположение, что в основе её лежат генетические аномалии, хотя способ передачи гена, который отвечает за заболевание, пока не доказан. В большинстве случаев каких-то изменений в хромосомном наборе не выявлено, хотя иногда бывают случаи, когда обнаруживаются раздробления хромосом. Поэтому точно говорить о генетической природе болезни пока можно с большой натяжкой.
Однако при наблюдении семьи, в которой есть дети с синдромом Корнелии де Ланге, можно обнаружить некоторые проявления заболевания и у родителей, и у других детей. Именно это даёт предположение о том, что болезнь может носить семейный характер.
Эпидемиология
Частота — 1:200000 новорожденных.
Клиническая картинка
Соматические нарушения: микроцефалия, брахицефалия, сросшиеся брови, загнутые ресницы, деформированные уши, маленький нос и неправильное его развитие, тонкая верхняя губа, высоко расположено нёбо, имеется расщелина, нарушения развития зубов, зрения, недоразвитие стоп и костей, сосков, мраморный оттенок кожи, повышенное количество волос на теле, судороги, низкий рост.
Внутренние органы: пороки развития сердца, половой системы, почек и др.
НПР: УО (разная степень), задержка развития, трудности в обучении, гиперактивность, нарушения внимания, агрессия, признаки аутизма,
Продолжительность жизни существенно сокращена.
Фенилкетонурия 1-го типа
Этиология
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией, которая вызывает снижение активности фермента фенилаланин-4-монооксигеназы, обеспечивающей превращение фенилаланина в тирозин. Фермент имеется только в печени, почках, поджелудочной железе.
Эпидемиология
Частота — 1:10000 новорожденных.
Клиническая картина
Первый признак — рвота, ребенок становится вялым, сонливым, не проявляется интерес к окружающему, раздражителен, беспокоен, плаксив, срыгивает, нарушение мышечного тонуса, судороги, повышенная потливость, «мышиный запах», отстают в физическом и нервно-психическом развитии
Продолжительность жизни в норме.
Мукополисахаридозы
Группа редких генетически заболеваний, причиной которых является нехватка определенных ферментов, которые помогают расщеплять определенные виды жиров и углеводов на простые молекулы.
Наследуется по аутосомно-рецессивному типу.
Выявляется непропорционально малый рост, задержка которого начинается к концу первого года жизни. Грубые черты лица: нависающий лоб, большой язык, гипертелоризм, деформация ушей и зубов. Грудная клетка деформирована, выражен кифоз (искривление назад) грудного и поясничного отделов позвоночника. Характерны ограничение подвижности суставов, увеличение селезенки и печени одновременно, пупочные и паховые грыжи. Раннее окостенение затылочно-теменного шва, «рыбьи позвонки» (позвонки как двояковогнутые линзы). Снижение мышечного тонуса, общая двигательная заторможенность. Снижение интеллекта и ослабление слуха разной степени характерны для различных типов мукополисахаридозов.
Мукополисахаридоз 1-го типа
Этиология
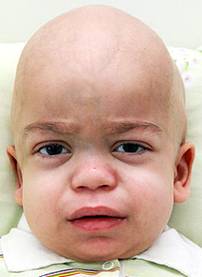 Аутосомно-рецессивное заболевание, обусловленное мутациями в структурном гене фермента альфа-L-идуронидазы, который расположен на коротком плече хромосомы 4.
Аутосомно-рецессивное заболевание, обусловленное мутациями в структурном гене фермента альфа-L-идуронидазы, который расположен на коротком плече хромосомы 4.
Эпидемиология
Частота — 1:100000 новорожденных.
Продолжительность жизни
Быстро прогрессирующее течение, приводящее к смертельному исходу на первом десятилетии жизни в результате сердечно-легочных и неврологических нарушений.
 43. Мукополисахаридоз 2-го типа
43. Мукополисахаридоз 2-го типа
Этиология
В основе лежат мутации Х-сцепленного гена идуронатсульфатазы, кодирующего одноимённый лизосомный фермент.
Эпидемиология
Частота — 1:140000-156000 новорожденных.
Клиническая картина
Соматические нарушения: макроцефалия, грубые черты лица, низкий рост, короткая шея, аномалии зубов, толстая кожа, атипичный пигментный ретинит (воспаление сетчатки) или разрушение сетчатки, грыжи, тугоподвижность суставов.
Внутренние органы: заболевания дыхательных путей, сердца
НПР: прогрессирующая проводящая или нейросенсорная потеря слуха, поражение ЦНС.
Продолжительность жизни
Дети с тяжелой формой погибают в конце второй декады жизни. Больные с легкой формой могут жить 50-60 лет.
Мукополисахаридоз 3-го типа
Этиология
Обусловлен дефицитом разных ферментов, но во всех случаях в лизосомах накапливается один тип гликозаминогликанов – гепарансульфат.Аутосомно-рецессивный тип наследования.
Эпидемиология
Частота — 1:70000 новорожденных.
Клиническая картина
Соматические нарушения: жесткие волосы, гирсутизм (рост волос по мужскому типу), отставание в росте, легкие скелетные изменения, выпячен живот.
Внутренние органы: увеличение печени и селезенки, пороки развития сердца
НПР: УО, регресс психомоторного и речевого развития, гиперактивны, аутичны или агрессивны, нарушен сна, небрежны и невнимательны, судороги.
Продолжительность жизни
Умирают в возрасте до 30 лет.
45. Факоматозы (общая характеристика)
Группа заболеваний, характеризующихся одновременным поражением кожи, нервной системы и внутренних органов, которые возникают из-за похожих эмбриональных нарушений.
В основе большинства факоматозов лежит генетически обусловленный дефект эмбрионального развития (мутация в одном из генов человека). Для каждой разновидности нейрокожных синдромов мутация локализуется в определенном гене.
Туберозный склероз
 Этиология
Этиология
Это генетическое заболевание с аутосомно-доминантным типом наследовани, характеризующееся развитием множественных гамартом и других опухолей.
Гамартомы – это доброкачественные опухоли, состоящие из тканей, аналогичных тем тканям и органам, в которых они развиваются. При этом структура их тканей является аномальной, отличающейся от нормы. Они могут располагаться в головном мозге, почках, коже, глазах.
Кариотип
Генетические дефекты могут быть расположены в области 9, 11, 16-й хромосом.
Эпидемиология
Частота — 1:5000 новорожденных.
Клиническая картина
Соматические признаки: изменения кожи и ее придатков, гипопигментированные (светлыми) пятна численностью от 3-4 до 100, обесцвечивание прядей волос, бровей, ресниц, аденомы сальных желез на лице (ангиофибромами лица), фиброзные уплотнения небольшого размера, создающие эффект шероховатости при ощупывании, околоногтевые фибромамы: тусклые множественные узелки на пальцах, под ногтями.
Внутренние органы: патология кишечника, глаз и др. внутренних органов.
НПР: УО (разная степень), эпилепсия.
Продолжительность жизни до 30 лет.
Синдром Стерджа-Вебера
 Этиология
Этиология
Это редкое врожденное заболевание, обусловленное нарушением эмбрионального развития, которое характеризуется возникновением сосудистых опухолей (ангиом) мягкой оболочки головного мозга и кожи лица (как правило, в области ветвей тройничного нерва: глазной и верхнечелюстной).Точные причины нарушения эмбрионального развития сосудистых сплетений не выявлены. Считается, что синдром передается по наследству, но не во всех случаях.
Эпидемиология
Частота — 1:50000 новорожденных.
Клиническая картина
Соматические признаки: сосудистые пытна (ангиомы) на лице, слабость в мышцах, снижение чувствительности на той стороне тела, где расположено пятно.
Внутренние органы: повышенное глазное давление или глаукомы, головная боль.
НПР: судороги, рассеянность, снижение памяти, задержка в умственном развитии, плаксивы, раздражительны, агрессивны.
Продолжительность жизни существенно не снижена.
Нейрофиброматоз 1-го типа
 Этиология
Этиология
Наследственное аутосомно-доминантное заболевание человека, обусловленное мутацией гена белка нейрофибромина, являющегося супрессором опухолевого роста, на 17 хромосоме. Заболевание характеризуется высокой пенетрантностью (частота появления) и высокой частотой возникновения новых мутаций.Мужчины и женщины поражаются одинаково часто.Причины неизестны.
Кариотип
Изменения в 17 хромосоме.
Эпидемиология
Частота — 1:3500-4000 новорожденных.
Клиническая картина
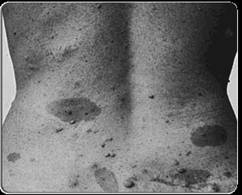
Соматические признаки: 6 и более пятен на коже цвета «кофе с молоком», каждое в диаметре более 5 мм у ребенка и более 15 мм у взрослого, 2 и более нейрофибромы (доброкачественных опухолей переферического нерва), усиленная окраска в подмышечной и паховой областях, 2 и более узелков Лиша - окрашенные гамартомы радужной оболочки глаза, выраженные костные аномалии.
Внутренние органы: глиома (опухоль) зрительного нерва, повышенный риск развития злокачественных опухолей.
НПР: эпилепсия, УО.
Продолжительность жизни без изменений.
 49. Нейрофиброматоз 2-го типа
49. Нейрофиброматоз 2-го типа
Этиология аналогично I типу.
Кариотип
Изменения 22 хромосомы.
Эпидемиология
Частота — 1:33000-50000.
Клиническая картина
Соматические признаки: проблемы со слухом и зрением, множественные образования а теле и лице.
Внутренние органы: двустороннее новообразование, вовлекающее слуховой нерв, односторонняя опухоль слухового нерва, звон в ушах, нарушение равновесия, высокий риск возникновения злокачественных опухолей.
НПР: судороги, УО.
Продолжительность жизни не изменена.
 50. Гипомеланоз Ито
50. Гипомеланоз Ито
 Этиология
Этиология
Врожденное заболевание, поражающее детей обоего пола и часто сочетающееся с дефектом других органов и систем.
Доказательств его наследственной передачи нет, но при нем описаны хромосомный мозаицизм и хромосомные транслокации.
Эпидемиология
Частота — 1:6000-7000 новорожденных.
Клиническая картина
Соматические признаки: потеря пигмента на различных участках тела, небольшие повреждения кожи, белые или бесцветные, линии Блашко, проблемы с зубами и ротовой полостью, зрением, ортопедические проблемы, патологии волос и ногтей.
Внутренние органы: сколиоз, заболевание почек, возможны опухоли ГМ.
НПР: судороги, задержка умственного развития, нарушения слуха, речи.
Продолжительность жизни не изменена.
 51. Миотоническая дистрофия
51. Миотоническая дистрофия
Этиология
Это самый распространенный тип мышечной дистрофии у взрослых. Заболевание передается по наследству аутосомно-доминантным путем. Дефектный ген локализован в хромосоме 19.
Кариотип
Изменения в 19 хромосоме.
Эпидемиология
Частота — 10:100000 населения.
Клиническая картина
Соматические признаки: слабость мышц лица, лицо удлиненное и худое, облысение в области лба, миотония (слабость мышц) конечностей, дегенерация мышц, нарушение походки, частые падения, постоянная усталость, приоткрытый рот.
Внутренние органы: блокада сердца, нарушение сердцебиения, тяжелые аритмии, отсутствие мышечных болей,
НПР: со временем утрачиваются физические навыки, УО.
Продолжительность жизни не сокращена. В конце концов становится совершенно беспомощным.
Миопатия Дюшенна
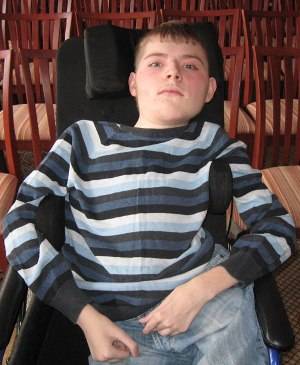 Этиология и кариотип
Этиология и кариотип
Тяжелое заболевание из группы наследственных прогрессирующих миопатий. В ее основе лежит наследственный дефект дистрофина - белка, поддерживающего целостность мембран мышечных волокон.
Болезнь наследуется рецессивно, сцепленно с Х-хромосомой, поэтому болеют мальчики. Женщины - носительницы гена не болеют. У большинства больных слабость становится заметной до трехлетнего возраста: они часто падают, с трудом поднимаются, постепенно утрачивают приобретенные двигательные навыки.
Эпидемиология
Частота — 3:10000 новорожденных мальчиков.
Клиническая картина
Соматические признаки: неустойчивая походка, неловкость в движении, постоянные падения при ходьбе, походка становится «утиной», не моет подняться по лестнице, трудности при подъеме из положения сидя и лежа, мнимая гипертрофия мышц (кажется, что мышцы здоровые и сильные, однако, это не так), с примерно 15 лет становится глубоким инвалидом, сколиоз, контрактуры суставов (конечность не может полностью согнуться/разогнуться)
Внутренние органы: перерождение мышечной ткани в жировую, поражение сердца и дыхания.
НПР: легкая УО.
Продолжительность жизни
Погибают к 20-летнему возрасту.
 53. Гемолитическая болезнь плода и новорожденного
53. Гемолитическая болезнь плода и новорожденного
Этиология
Это состояние, вызванное конфликтом между эритроцитами матери и плода. Патология наблюдается в тех случаях, когда у женщины имеется отрицательный резус или конфликт по группе крови.
Эпидемиология
Частота, связанная с резус-несовместимостью, возрастает с каждыми последующими родами.
Клиническая картина
Первый вариант – водянка плода. Развивается под долгим воздействием антител на плод. Проявления этого заболевания видны при рождении: увеличенный живот (в связи с увеличенной печенью и селезенкой), ярко выраженные отеки, так же на лицо все признаки угнетения нервной системы. Кожа у новорожденного будет бледного оттенка, могут проступить кровоподтеки подкожные. Синдром дыхательного расстройства.
Второй вариант: желтуха, желтушность кожи, увеличение селезенки и печени, слабо выраженная отечность. На лицо все признаки анемии: бледность слизистых оболочек и кожных покровов.
Третий вариант – ядерная желтуха. Этот вариант болезни обусловлен тем, что непрямой билирубин проникает в ткани головного мозга. На третьи или четвертые сутки после рождения ребенка у него появляются все признаки поражения мозговой системы, которые ведут к неврологическим нарушениям.
Продолжительность жизни
Зависит от варианта заболевания.
Микроцефалия
 Этиология
Этиология
Патология развития головного мозга. Синдром микроцефалии заключается в значительном уменьшении массы мозга и соответственном недоразвитии черепа. Черепная коробка формируется неестественно маленькой, отклоняясь при развитии на 2 и более сегмента.
Возникает спорадически. Зависит от многих причин (от окружающей среды до образа жизни матери). Наследуется аутосомно-рецессивно.
Эпидемиология
Частота — 1:1000 новорожденных.
Клиническая картина
Соматические признаки: голова меньше нормы, маленький родничок, быстро закрывается, узкий покатый лоб, оттопыренные уши, выпуклые надбровные дуги, низкий мышечный тонус, вес и рост ниже нор







