Концентрация свободной глюкозы в клетках значительно выше ее содержания во внеклеточном секторе. Известно, что скорость переноса глюкозы через наружную клеточную мембрану мио- и адипоцитов представляет собой детерминанту интенсивности фосфорилирования глюкозы в клетках. D-глюкоза и другие сахара проникают в клетки путем облегченной диффузии, опосредованной переносчиком. Инсулин усиливает трансмембранный перенос глюкозы, увеличивая число переносчиков, активно функционирующих на уровне плазматической мембраны. Недостаточная концентрация нормального инсулина в циркулирующей крови, нарушения взаимодействия гормона со своим рецептором приводят к снижению числа переносчиков глюкозы, функционирующих на уровне плазматической мембраны. В результате падает транспорт глюкозы в клетки жировой и мышечной ткани, что повышает содержание глюкозы во внеклеточном пространстве и обуславливает гипергликемию.
Инсулин не усиливает облегченной диффузии глюкозы в гепатоци-ты, но увеличивает приток глюкозы в дифференцированные клетки печени через рост активности в них глюкокиназы, одного из ключевых ферментов гликолиза, превращающего глюкозу в глюкозо-6-фосфат. Инсулин, стимулирующий экспрессию гена данного энзима, повышает содержание глюкокиназы в гепатоцитах, что интенсифицирует гликолиз и снижает содержание свободной глюкозы в клетках печени. В резуль-
тате растет свободная диффузия глюкозы из внеклеточного сектора в гепатоциты. При инсулинопении или патологической резистентности по отношению к инсулину на уровне гепатоцитов в них падает фосфорили-рование глюкозы. В результате растет содержание свободной глюкозы в дифференцированных клетках печени и падает ее свободная диффузия в гепатоциты. Это повышает содержание глюкозы во внеклеточном секторе и служит одним из факторов гипергликемии у больных сахарным диабетом.
Кроме того, инсулин усиливает интенсивность гликолиза в печени и других органах вследствие того, что повышает активность в них других его ферментов, фосфофруктокиназы и пируваткиназы. Одновременно инсулин подавляет в гепатоцитах активность глюкозо-6-фосфатазы, что повышает концентрацию в них глюкозо-6-фосфата. В результате действия инсулина глюкоза удерживается в клетках печени, так как их наружная мембрана непроницаема для глюкозо-6-фосфата. Снижение содержания инсулина в крови и (или) патологическая рези-стентность по отношению к инсулину через повышение активности глюкозо-6-фосфатазы снижают удержание глюкозы в гепатоцитах в виде глюкозо-6-фосфата. Это может быть одним из факторов гипергликемии. (
Инсулин, повышая активность гликогенсинтетазы, усиливает образование гликогена на системном уровне. Поэтому инсулинопения и патологическая резистентность по отношению к инсулину снижают утилизацию глюкозы из внеклеточного сектора для синтеза гликогена и тем самым обуславливают гипергликемию.
Под влиянием инсулина происходит избирательное ингибирование транскрипции гена, кодирующего фосфоенолпируваткарбоксикиназу, ключевой фермент глюконеогенеза. В результате инсулинопении или патологической резистентности к инсулину глюконеогенез растормаживается, что повышает высвобождение глюкозы печенью и ведет к гипергликемии.
Итак, патогенез гипергликемии у больных сахарным диабетом весьма сложен и не сводится только к низкой облегченной диффузии глюкозы через наружную клеточную мембрану клеток мышечной и жировой ткани (схема. 13.1). Свою роль в развитии гипергликемии у пациентов с сахарным диабетом играют превалирующие на системном уровне эффекты катаболических гормонов антагонистов инсулина в виде торможения синтеза гликогена и усиления гликогенолиза.
Инсулинопения и патологическая резистентность по отношению к инсулину через снижение анаболического эффекта инсулина и его анти-катаболического действия обуславливают стойкое падение процессов анаболизма, в том числе липогенеза и белкового синтеза. Одновременно
в мышечные и другие клетки вследствие недостаточного системного эффекта инсулина падает транспорт аминокислот, катионов калия и кальция, нуклеозидов и органического фосфата как субстратов анаболизма. Снижение белкового синтеза у больных сахарным диабетом захватывает и систему иммунитета, что служит одной из причин иммунодефицита, вторичного по отношению к сахарному диабету. Падение анаболизма и связанная с ним интенсификация липолиза и распада белков у больных инсулинзависимым сахарным диабетом, возникшим в детском возрасте, может обусловить задержку роста тела. После возникновения инсулинзависимого сахарного диабета у взрослых болезнь приводит к снижению массы тела.

Схема 13.1. Патогенез гипергликемии у больных сахарным диабетом
Предположительно инсулин влияет на образование атерогенных ли-попротеинов или снижает очищение от них плазмы крови. Очевидно падение этого эффекта гормона у больных сахарным диабетом приводит к ускоренному атеросклерозу.
Морфопатогенез атеросклероза у больных сахарным диабетом не отличается от патогенетических механизмов атеросклеротических патологических измененеий сосудистой стенки у пациентов, не страдающих от сахарного диабета. И в том и в другом случае патогенные межклеточные взаимодействия в основном складываются из аномального изменения функционального состояния эндотелиоцитов, адгезии к ним активированных мононуклеарных фагоцитов сосудистой стенки, за которой
следует прилипание агрегатов активированных тромбоцитов. Агрегаты этих активированных клеток становятся источником цитокинов со свойствами факторов клеточного роста, что обуславливает пролиферацию гладкомышечных миоцитов сосудистой стенки как этапа формирования атеросклеротической бляшки. Многие из факторов риска атеросклероза, гиперхолестеринемия, патогенно низкая концентрация в крови липопро-теинов высокой плотности, курение и артериальная гипертензия одновременно представляют собой факторы риска сахарного диабета второго типа. Факторы риска атеросклероза, связанные исключительно с сахарным диабетом, - это гипергликемия, гиперинсулинемия (патологически высокая концентрация инсулина в крови у больных сахарным диабетом второго типа, преимущественно обусловленным патологической резистентностыо^гю отношению к инсулину), а также гипертриг-лицеридемия.
Патогенез диабетической микроангиопатии
В настоящее время не вызывает сомнений, что именно гипергликемия является основной причиной патологических изменений сосудов у больных с длительным инсулинзависимым сахарным диабетом. Тем не менее, патогенез диабетической ангиопатии вследствие гипергликемии остается неясным. Так как глюкоза и ее метаболиты выступают субстратами на различных путях метаболизма, то ангиопатический эффект гипергликемии реализуется несомненно через действие множества патогенетических механизмов. В последние десятилетия были предложены четыре теоретических модели действия таких механизмов:
» связанный с активностью альдозоредуктазы (сорбитоловый путь);
«через изменение активности протеинкиназы С;
» неферментного гликолизирования;
» в результате нарушений окислительно-восстановительного потенциала тканей.
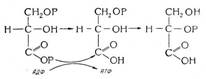
В большинстве тканей организма глюкоза может трансформироваться в сорбитол через активность альдозоредуктазы. Из-за высокой константы Михаэлиса' альдозоредуктазы для глюкозы уровень синтеза сорбитола при низкой концентрации углевода в клетках невысок. При
Константа Михаэлиса - концентрация субстрата, при которой скорость реакции, катализатором которой является данный фермент, составляет половину максимальной (в данном случае субстрат - это глюкоза, фермент - альдозоре-Дуктаза, а реакция - это образование сорбитола из глюкозы).
гипергликемии растет содержание глюкозы в клетках, в которые углевод проникает путем обычной, а не облегченной диффузии, усиливаемой инсулином. В результате в клетках усиливается образование сорбитола. После образования сорбитола возможна его метаболизация, связанная с активностью сорбитолдегидрогеназы, в результате которой образуются фруктоза и восстановленный никотинамидадениндинуклеотид (NADH). Однако превращение сорбитола во фруктозу и NADH идет медленно, и сорбитол накапливается в клетках. Рост содержания сорбитола в клетках (в частности в нейронах) приводит к их отеку вследствие миграции воды из внеклеточного сектора в сторону большей осмотической концентрации сорбитола.
Предполагают, что накопление сорбитола в клетках хрусталика глаза больного инсулинзависимым сахарным диабетом приводит к образованию у таких пациентов диабетической катаракты.
Так как возрастание концентрации сорбитола в нейронах и клетках мышечной ткани не может привести к их значительному отеку и обусловить цитолиз, то аккумуляцию сорбитола нельзя признать ведущим механизмом диабетических ангио- и нейропатий.
Полагают, что связанный с инсулинопенией рост в клетках и тканях концентрации свободной глюкозы и сорбитола может быть причиной увеличения содержания в них миоинозитола как причины диабетической ангиопатии. Это может быть только предположением, ибо известно, что рост концентрации миоинозитола вследствие инсулинопении, гипергликемии и высокой активности альдозоредуктазы преимущественно происходит в клетках нервной ткани. В эксперименте ингибиторы альдозоредуктазы снижают содержание миоинозитола только в нейронах. Одновременно с ростом содержания в нейронах миоинозитола в них снижается активность натрий-калий-АТФазы. Полагают, что снижение активности натрий-калий-АТФазы также представляет собой следствие накопления в нейронах сорбитола, так как ее активность восстанавливается под влиянием ингибиторов активности альдозоредуктазы.
Были получены обнадеживающие результаты экспериментального изучения ингибиторов альдозоредуктазы как средств коррекции диабетических ангио- и нейропатий. Тем не менее, клинические испытания не принесли ожидаемых результатов, которые бы свидетельствовали о возможности предупреждения диабетических ретино- и нейропатий через действие ингибиторов альдозоредуктазы.
Не исключено, что гипергликемия вызывает диабетическую микро-ангиопатию через патологические изменения обмена фосфолипидов и активности протеинкиназы С. Некоторые из фосфолипидов и данный фермент, функционируя в качестве агентов системной и паракринной
регуляции, оказывают на функциональное состояние сосудов и его характеристики (проницаемость сосудистой стенки, сократимость ее глад-комышечных элементов, экспрессия тромбогенного потенциала эндоте-лиоцитов, объемная скорость кровотока, внутренняя секреция, синтез составляющих базальной мембраны) широкий спектр в том числе и разнонаправленных влияний. Иными словами, данные системы регуляции клеток на уровне сосудистой стенки контролируют те функции ее клеточных элементов, расстройства которых и служат причинами диабетической ангиопатии. Предположительно повышение активности протеинкиназы С, вызывающее патогенную разностороннюю модуляцию функционального состояния микрососудов, может быть ведущим звеном патогенеза диабетической микроангиопатии. В пользу этого предположения говорит факт высокой активности протеинкиназы С в сетчатой оболочке глаз пациентов,—страдающих от инсулинзависимого сахарного диабета и диабетической ретинопатии; кроме того, известно, что у таких пациентов активность данного фермента аномально высока в стенке аорты, кардиомиоцитах, а также в почечных клубочках. Экспериментальные исследования сахарного диабета показали, что инсулинопения и гипергликемия ведут к росту активности протеинкиназы С в стенках сосудов сетчатой оболочки глаза, аорты, почечных клубочков и венечных артерий. Одновременно с ростом активности энзима в клеточных элементах сосудистой стенки определяли возрастание уровня содержания диацилглицерола.
Это позволяет предположить, что инсулинопения и гипергликемия повышают активность протеинкиназы через увеличение содержания в клетках диацилглицерола, одна из функций которого - это усиление перемещения протеинкиназы С из неактивного пула фермента в цитозоле в его активный мембранный пул. Полагают, что изменения активности протеинкиназы С, связанные с сахарным диабетом и гипергликемией, происходят преимущественно в сосудистой стенке, где вследствие инсулинзависимого сахарного диабета растет активность специфической для сосудистой стенки бета-П-протеинкиназы С. В основном рост активности протеинкиназы С меняет функциональное состояние сосудистой стенки посредством модуляции экспрессии генов эндотелиаль-ных и других клеток сосудистой стенки.
Данные, полученные при экспериментальном исследовании влияний гипергликемии на обмен веществ, свидетельствуют, что рост активности протеинкиназы С вследствие гипергликемии не связан с изменением осмоляльности внеклеточной жидкости и со сдвигами в обмене сорбитола.
Обусловленные гипергликемией рост содержания диацилглицерола и активности протеинкиназы С при инсулинзависимом сахарном диабе-
те возникают не сразу и не сразу подвергаются обратному развитию после устранения гипергликемии. Тут мы имеем дело еще с одним проявлением общей для развития патологических процессов закономерности, то есть тенденции к эндогенизации - относительной потери связи источников их развития с первичным причинным фактором. Причина эндогенизации в данном случае состоит в том, что протеинкиназа С, комплексно меняя экспрессию генома клетки формирует патологическую систему взаимодействия элементов генетического материала клетки как причины персистирования диабетической ангиопатии (это всего лишь наше предположение — В.Ю. Шанин).
Полагают, что гипергликемия вызывает патогенные межклеточные взаимодействия как причину диабетической ангиопатии через образование соединений на основе ковалентных связей глюкозы с ингредиентами клеточных мембран и (или) циркулирующими с кровью липопротеина-ми и белками. Процесс образования таких соединений называют неферментным гликолизированием. Через соединение глюкозы ковалентными связями с аминогруппами протеинов клеточных мембран, ядерных белков и дезоксирибонуклеиновой кислоты происходит образование так называемых транзиторных промежуточных продуктов неферментного гликолизирования. Эта реакция достигает своего равновесия за несколько недель. В дальнейшем промежуточные продукты неферментного гликолизирования служат субстратами для образования конечных продуктов неферментного гликолизирования. С образованием конечных продуктов неферментного гликолизирования реакция становится необратимой. Конечные продукты неферментного гликолизирования могут обуславливать патогенную модуляцию экспрессии генома клеток сосудистой стенки как причину диабетической микроангиопатии. Кроме того, они служат субстратами образования свободных кислородных радикалов, которые повреждают наиболее в функциональном отношении активные фосфолипиды клеточных мембран и еще в большей степени стабилизируют конечные продукты неферментного гликолизирования.
Конечные продукты неферментного гликолизирования образуются во внеклеточном пространстве через соединение глюкозы ковалентными связями с липопротеинами низкой плотности, альбумином и гемоглобином. Образование конечных продуктов неферментного гликолизирования на поверхности клеточных мембран меняет их механические свойства, повышая жесткость мембраны. В результате плазматическая мембрана эритроцитов становится более ригидной, что снижает деформируемость эритроцита как необходимое условие нормальной микроциркуляции (незатрудненного прохождения эритроцитов по микрососудам и капиллярам). Эритроциты с патологически низкой вследствие гипер-
гликемии и неферментного гликолизирования деформируемостью при прохождении по микрососудам активируют эндотелиальные клетки через усиленное трение о часть их наружной мембраны, обращенной в просвет микрососуда. Активация эндотелиоцитов служит первым этапом патогенных межклеточных взаимодействий, приводящих к диабетической микроангиопатии.
Кроме того, звеном патогенеза диабетической микроангиопатии является утолщение базальной мембраны капилляров в результате образования входящим в ее состав коллагеном и глюкозой конечных продуктов неферментного гликолизирования, при котором молекулярные цепочки, составляющие коллаген, соединяются между собой попереречными связями в виде молекул глюкозы. В результате базальная мембрана не только утолщается. В ней падает содержание протеогликанов, а функционирование интегриновых рецепторов сосудистой стенки меняется таким образом, что возникают дисфункции всех ее клеточных элементов.
Интенсивность образования конечных продуктов неферментного гликолизирования представляет собой прямую функцию длительности гипергликемии и степени патологического возрастания концентрации глюкозы в циркулирующей крови.
При длительной гипергликемии конечные продукты неферментного гликолизирования (КПНфГ) начинают циркулировать с кровью и фиксируются на наружной1поверхности ее мононуклеарных фагоцитов, которая содержит рецепторы специфические по отношению к КПНфГ. В результате происходит системная активация мононуклеарных фагоцитов, циркулирующих с кровью. Активированные мононуклеары начинают высвобождать такие цитокины как фактор некроза опухолей, ин-терлейкин-1, что приводит к гиперцитокинемии. Гиперцитокинемия через системные активацию эндотелиоцитов и рост экспрессии их тромбо-генного потенциала вызывает диабетическую микроангиопатию на уровне всего организма.
Если атеросклероз считать результатом хронического воспаления сосудистой стенки, основным клеточным эффектором которого являются активированные клетки системы мононуклеарных фагоцитов, то ги-перцитокинемию вследствие активации мононуклеаров в результате воздействия на них КПНфГ можно предположительно признать фактором ускорения атеросклероза у больных сахарным диабетом и диабетической микроангиопатией.
Длительная гипергликемия и неферментное гликолизирование приводят к образованию связей в виде молекул глюкозы, связанных с аминогруппами, между цепочками нуклеотидов дезоксирибонуклеиновой кислоты (ДНК). Это вызывает дисфункции генетического материала эндотелиоцитов, обусловленные изменением строения двойной спирали
ДНК, что предположительно можно считать еще одним фактором диабетической микроангиопатии.
В эксперименте было показано, что инфузия растворов, содержащих конечные продукты неферментного гликозилирования, вызывает характерные для диабетической микроангиопатии утолщение базальной мембраны микрососудов и патологические изменения сократимости глад-комышечных элементов сосудистой стенки. При этом ослабляется физиологическая реакция расширения сосудов в ответ на действие такого медиатора как оксид азота. В этой связи были сделаны предположения о механизме ускоренного развития тяжелой первичной артериальной ги-пертензии у больных сахарным диабетом и длительной гипергликемией, который состоит в потере гладкомышечными клетками сосудистой стенки нормальной реактивности по отношению к расширяющему сосуды действию оксида азота.
Обусловленный гипергликемией рост утилизации глюкозы как субстрата гликолиза или образования полиола увеличивает отношение содержания в клетке восстановленной формы никотинамидадениндинук-леотида к его окисленной форме (NADH /NAD). Предполагают, что рост содержания в клетке NADH представляет собой следствие интенсификации обмена сорбитола и утилизации глюкозы в реакциях пентозного шунта, что в частности приводит к образованию 1,3-дифосфоглицерата. Рост NADH /NAD патогенно влияет на клеточные функции через патологические изменения а)синтеза диацилглицерола, б)функционирования систем восстановления структуры дезоксирибонуклеиновой кислоты и в) окисления жирных кислот. В эксперименте было продемонстрировано обратное развитие диабетической микроангиопатии под влиянием пирувата как метаболита, снижающего NADH /NAD.
Самым ранним из гистопатологических сдвигов при диабетической ретинопатии является потеря капиллярами сетчатой оболочки своих перицитов. За несколько лет хронической гипергликемии соотношение между эндотелиальными клетками и перицитами ретинальных микрососудов может снизиться от нормального 1:1 до патологического 1:10. Кроме того, диабетическую микроангиопатию на уровне сетчатой оболочки глаза характеризуют расширение капилляров, утолщение их базальной мембраны, рост проницаемости и возникновение капиллярных микроаневризм. Все эти патологические изменения капилляров сетчатой оболочки можно связать с утратой нормальных межклеточных взаимодействий между перицитами и эндотелиальными клетками. Известно, что гипергликемия угнетает клеточный рост перицитов. Перициты оказывают на эндотелиальные клетки регуляторные влияния, которые служат необходимым условием нормальной экспрессии эндотелиоцитами цитокинов. В результате потери перицитов экспрессия генома и клеточ-
ный рост эндотелиоцитов претерпевают патологические изменения. Это обуславливает потерю эндотелиоцитами способности обеспечивать нормальный кровоток в капиллярах. В результате часть капилляров оказывается обтурированными микротромбами и появляются капилляры, не содержащие крови («капилляры-призраки»). Вторичная микроциркулятор-ная гипоксия приводит к локальному росту концентрации соответствующих факторов клеточного роста, что обуславливает разрастание капилляров как причину пролиферативной диабетической микроангиопатии на уровне сетчатой оболочки глаза (пролиферативная ретинопатия).
Пролиферативную ретинопатию характеризует прорастание новообразуемых капилляров в стекловидное тело. Новые капилляры неполноценны. Число окружающих их перицитов резко снижено. В результате стенка капилляров теряет свои нормальные свойства барьера между кровью и интерстицием, что служит причиной множества кровоизлияний в сетчатую оболочку, ее отслойки и потери зрения Из факторов клеточного роста и цитокинов, вовлеченных в патогенез пролиферативной ретинопатии, следует выделить следующие: основные и кислые факторы роста фибробластов, инсулиноподобный фактор роста I, бета трансформирующий фактор клеточного роста, а также сосудистый эндотелиаль-ный фактор клеточного pocta.
Гипертрофия мезангиалвдых клеток почечных клубочков у больных сахарным диабетом во многом составляет диабетическую нефропатию. В результате пролиферации клеток мезангия клубочки теряют фильтрационную поверхность, формируемую эндотелиальными и эпителиальными клетками. При этом утолщается базальная мембрана и теряются эндотелиальные и эпителиальные клетки. Все это завершается склерозом почечных клубочков и чреватым танатогенезом угнетением экскреторных функций почек. При диабетической нефропатии в клубочках растет содержание таких факторов клеточного роста как альфа-фактор некроза опухолей, бета трансформирующий фактор клеточного роста, фактор клеточного роста тромбоцитов, а также основной фактор роста фиброластов.
Развитие диабетической нефропатии и гипертрофию мезангиальных клеток связывают с ростом содержания в циркулирующей крови ангио-тензина II. При этом полигенную наследственную предрасположенность к развитию диабетической нефропатии у больных сахарным диабетом связывают с полиморфизмом генов ангиотензинпревращающего фермента и ангиотензиногена. Максимальный уровень риска диабетической нефропатии и соответственно предрасположенности к ней у больных сахарным диабетом фиксируют тогда, когда ген ангиотензинпревращающего фермента содержит два аллеля D, а ген ангиотензиногена составляют два одинаковых аллеля, экспрессия которых дает молекулу
ангиотензиногена, в которой на 235 позиции находится треонин. Кроме того, свою лепту в предрасположенность к диабетической нефропатии вносит полиморфизм гена рецептора ангиотензина II, который предположительно содержит наружная мембрана клеток мезангия. Если вспомнить о том, что определенная системная гиперактивация эндотелилаль-ных клеток, участвующих в образовании ангиотензина II, во многом составляет патогенез осложнений сахарного диабета и, то можно считать диабетическую микроангиопатию инициирующим моментом роста образования ангиотензина II, который происходит особенно интенсивно при соответствующем полиморфизме генов.
Образование конечных продуктов неферментного гликолизирования миелина предположительно лежит в основе диабетической нейропатии, которая в начале развития захватывает тонкие нервные волокна. Если оценивать вагальные регуляторные влияния на сердце, фиксируя вариабельность частоты сердечных сокращений при глубоком дыхании и ориентируясь на возрастную норму, то у большинства больных инсулинза-висимым сахарным диабетом можно выявить признаки блокады вагаль-ных регуляторных влияний на сердце, которая приводит к меньшей вариабельности. С возрастом вариабельность падает. Снижение вариабельности частоты сердечных сокращений при глубоком дыхании у больных инсулинзависимым сахарным диабетом происходит в несколько раз быстрее. Данные полученные при исследовании вариабельности частоты сердечных сокращений при глубоком дыхании свидетельствуют, что диабетическая нейропатия развивается у большинства из них, но только у небольшой части таких пациентов она приводит к заметным и тяжелым последствиям нарушений вегетативной регуляции функций:
» ощущению жара и боли в конечностях;
«патологически интенсивному потоотделению;
» задержке мочеиспускания;
» расстройствам сердечной проводимости со снижением интервала QT, сердечным аритмиям;
» диареям и запорам;
«угнетению моторики желудка как причины застоя желудочного содержимого и рвоты.