14.1. Двухстадийная реакция гомогенного катализа при одной лимитирующей стадии
По современным представлениям катализом называют изменение скорости химической реакции при воздействии вещества-катализатора, которое многократно участвует в элементарных химических реакций, но не входит в состав продуктов и не находится в стехиометрических отношениях с ними. Катализатор регенерируется после каждого цикла превращения реагентов в продукты и может расходоваться только по побочным реакциям.
В типичных случаях гомогенного катализа вначале по обратимой реакции образуется активный комплекс катализатора К с одним из реагентов (субстратом) А, причем эта стадия состоит в присоединении или замещении:

Затем образовавшийся комплекс (или активная частица) по бимолекулярной реакции взаимодействует со вторым реагентом или по мономолекулярной реакции распадается, с образованием молекулы продукта и регенерацией катализатора:

Возьмем для кинетического анализа наиболее общий случай, когда первая стадия состоит в замещении, а вторая является бимолекулярным взаимодействием. Скорость реакции описывается кинетическим уравнением:

где k2 ‒ истинная константа скорости каталитической реакции.
Концентрацию активного комплекса (или частицы) находим из условия стационарности
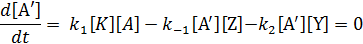
откуда

Подставляем значение 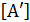 в уравнение скорости реакции и получаем:
в уравнение скорости реакции и получаем:

Если первая стадия сводится к присоединению, [Z] в уравнении исчезает, превращаясь в единицу. При мономолекулярности второй стадии то же происходит с [Y]. Аналогичные изменения наблюдаются, когда эти вещества находятся, в большом избытке или являются растворителями, что позволяет ввести, их концентрации в соответствующие константы скорости.
При анализе уравнения скорости представляют интерес два предельных случая:
1. Скорость превращения промежуточного комплекса на второй стадии значительно превышает скорость его обратного разложения на исходные вещества,, т., е. k 2[Y] >> k -1[Z]. В этом случае величиной k -1[Z] в знаменателе можно пренебречь, а комплекс не может накопиться в заметном количестве. Если другие формы состояния веществ отсутствуют, то [К] = СK и [А] = СА, что дает следующее уравнение для скорости реакции

Реакция имеет первые порядки по катализатору и реагенту первой стадии, нулевок порядок по реагенту второй стадии и не тормозится веществом Z. Ее константа скорости равна константе скорости элементарной стадии образования промежуточного комплекса (k 1), которую легко найти, разделив экспериментально найденную эффективную константу k эфна концентрацию катализатора.
Следовательно, в данном случае кинетическое уравнение включает характеристики только первой стадии процесса, которая оказывается лимитирующей.
2. Скорость второй стадии реакции значительно меньше скорости обратного превращения промежуточного комплекса в исходные реагенты, т. е. k2 [Y]<< k-1 [Z]. Это позволяет пренебречь величиной k2 [Y]в знаменателе и уравнение 14.1 принимает вид:
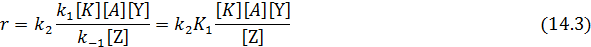
В кинетическое уравнение (14.3) входят константа скорости второй стадии и лишь константа равновесия первой, т.е. общая скорость процесса лимитируется второй стадией ‒ превращением промежуточного комплекса в продукты.
Равновесная концентрация промежуточного комплекса может достичь значительной величины, и тогда аналитическая концентрация катализатора будет суммой:

откуда
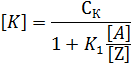
Если реагенты находятся в избытке, можно принять, что [А] ≈ СА, [Z] = СZ и [Y] = СY, то кинетическое уравнение (14.3) принимает вид:

При очень малой константе равновесия можно пренебречь в знаменателе слагаемым К1СА, что дает:

Если первая стадия состоит в присоединении или вещество Z является растворителем и его концентрация входит в выражение для константы, получим:

Оно становится подобным по форме с (14.2), и два разных механизма оказываются кинетически неразличными. Для решения вопроса о лимитирующей стадии приходится прибегать к другим способам, например к определению энтропии активации или реакционной способности, кинетическому изотопному методу и др.
Если константа равновесия при образовании промежуточного комплекса достаточно велика, слагаемым К 1САв знаменателе уравнения уже пренебречь нельзя. В отсутствие вещества Z (при первой стадии присоединения) или при наличии его в большом избытке (растворитель) уравнение дает:

Если Y является растворителем или если вторая стадия мономолекулярна, из уравнения (14.5) выпадает СY. При очень большой константе К 1СА иногда можно пренебречь в знаменателе единицей по сравнению с К 1СА, в результате чего получим:

Оказывается, что в этом случае реакция имеет нулевой порядок по реагенту А. Физический смысл этого состоит в том, что ввиду почти количественного образования промежуточного комплекса дальнейшее повышение концентрации А уже не ведет к ускорению реакции.
Из уравнений видно, что даже для одной и той же реакции может наблюдаться разная зависимость скорости от концентрации реагента А. При малой ее величине, если можно пренебречь К1СА по сравнению с СZ или единицей, имеем линейную зависимость (первый порядок по А). Наоборот, при высокой СА, если можно пренебречь СZ или единицей в знаменателе, получим, что скорость не зависит от концентрации этого реагента (нулевой порядок по А). В промежуточной области концентраций будет наблюдаться постепенно замедляющийся рост скорости.
 Рис. 14.1. Зависимость скорости гомогенно-каталитической реакции от концентрации реагента А
Рис. 14.1. Зависимость скорости гомогенно-каталитической реакции от концентрации реагента А
| Эти закономерности изображены на рис. 14.1, причем по горизонтальному участку кривой легко находится истинная константа скорости каталитической реакции (k 2), равная эффективной константе, деленной на концентрацию катализатора.
Рассмотренный тип кинетических уравнений со второй лимитирующей стадией и значительной концентрацией промежуточного комплекса также нередко встречается в органическом синтезе.
|
Так, при взаимодействии α‒окисей с кислотными реагентами (фенолами, карбоновыми кислотами) и нуклеофильном катализе сопряженными основаниями комплекс α‒окиси с реагентом образуется в значительном количестве, и реакция слабо ускоряется с повышением концентрации фенола или карбоновой кислоты.
Другой пример ‒ эпоксидирование аллилового спирта перекисью водорода в присутствии вольфрамовой кислоты, которая быстро окисляется перекисью в надвольфрамовую кислоту, и весь катализатор практически находится только в этой форме. В результате реакция имеет нулевой порядок по перекиси водорода и первый ‒ по катализатору и аллиловому спирту в соответствии с уравнением (14.6):

Встречаются и такие реакции, когда катализатор находится в большом избытке, а концентрация реагента, наоборот, незначительна (сульфирование и сульфатирование слабо растворимых в серной кислоте веществ, нитрование смесями серной и азотной кислот и др.). Здесь можно принять, что [К] ≈ СK и [Y] ≈ СY, а концентрация А определяется выражением:

Подставляя находимую из этого уравнения величину [А] в уравнение скорости, получим:

В этом случае имеется переменная зависимость скорости (аналогичная изображенной на рис. 14.1) от концентрации не реагента, а катализатора, причем по горизонтальному участку кривой можно вновь определить истинную константу скорости каталитической реакции.
14.2. Кислотно-основный катализ
К кислотно-основному катализу применимы изложенные выше закономерности, в то же время имеется ряд особенностей.
В современной химии используются две теории кислот и оснований: протонная теория Брёнстеда-Лоури и электронная теория Льюиса.
14.2.1. Кислоты и основания Брёнстеда
По Брёнстеду-Лоури кислотами называют вещества, способные поставлять (отдавать) протон, а основания – вещества, способные присоединять протон:

Кислота и основание в растворе взаимосвязаны сопряженным процессом переноса протона. В растворе протон не существует в свободном виде и находится только в сольватированном состоянии (например, в воде Н3О+, Н5О2+, Н7О3+). Кислотный характер соединения (АН или ВН+) может проявляться только в присутствии основания (В:, А–) или растворителя (SН), обладающего основными свойствами, и наоборот, основный характер соединения (В:, А–) может проявляться исключительно в присутствии кислоты (АН, ВН+) или растворителя (SН), обладающего кислотными свойствами:

Таким образом, протонная кислота (основание) находятся в реакционной массе в нескольких формах: недиссоциированной кислоты АН (или сопряженного основания В:), иона лиония SH2+ (или лиата S-), а также в виде протонированных форм реагентов и продуктов. Кроме того сам растворитель SH может обладать кислотными или основными свойствами.
Диссоциация кислот и оснований – сложный процесс, который зависит от растворителя: диэлектрической проницаемости, сольватирующей способности и способности выступать в качестве кислот или оснований. Следовательно, кислотность и основность вещества в растворе зависят от основности (кислотности) растворителя.
Упрощенно кислотно-основное (протолитическое) равновесие можно представить схемой:

где А– Н···:В – комплекс с водородной связью;
А–···Н+–В- ‒ионная пара (или сольвато-разделенная электронная пара);
А– и ВН+ ‒ свободно сольватированные ионы.
Аналогично В: можно рассматривать и растворитель SН.
Полный перенос протона от кислоты к основанию по Брёнстеду реализуется лишь в исключительных случаях, когда в кислотно-основном равновесии участвуют сильные кислоты и основания в высокополярных растворителях. Обычно при кислотно-основном взаимодействии наблюдается частичный перенос протона от кислоты АН к основанию или растворителю, а в ряде случаев проходят все стадии, описанные выше приведенной схемой.
Если кислотно-основное равновесие между кислотой ирастворителем SH может быть описано схемой Брёнстеда:

то тогда термодинамическая константа протолитического равновесия запишется выражением

где  ‒ активности соответствующих частиц в растворе;[А–], [АН], [SH], [SH2+] ‒ концентрации соответствующих частиц в растворе;
‒ активности соответствующих частиц в растворе;[А–], [АН], [SH], [SH2+] ‒ концентрации соответствующих частиц в растворе;
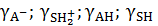 ‒ коэффициенты активности соответствующих частиц в растворе; Кс ‒ концентрационная константа равновесия.
‒ коэффициенты активности соответствующих частиц в растворе; Кс ‒ концентрационная константа равновесия.
Термодинамические константы кислотности К аАНи К аSН2 +, характеризующие силу кислот АН и SH2+ независимо от среды, относятся к соответствующим протолитическим кислотно-основным процессам как результат двух сопряженных реакций:


Константа протолитического равновесия К равна отношению термодинамических констант кислотности сопряженных кислот:

Таким образом, можно измерить величину К, характеризующую протолитическое равновесие реакции, но нельзя определить абсолютные значения 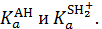 Константу кислотности можно вычислить только относительно стандарта. В качестве последнего выбирают растворитель SH, отвечающий паре сопряженной кислоты и основания: SH2+ - SH. Например, для водных растворов Н3О+ - Н2О.
Константу кислотности можно вычислить только относительно стандарта. В качестве последнего выбирают растворитель SH, отвечающий паре сопряженной кислоты и основания: SH2+ - SH. Например, для водных растворов Н3О+ - Н2О.
В сильно полярном растворителе, например в воде (ε = 78,5), при определенной температуре:



Так как в разбавленных растворах, в данном случае в воде, количество растворителя ‒ величина практически постоянная, константу равновесия "стандартной" реакции можно принять равной единице:
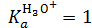 , тогда
, тогда 
Константу кислотности кислоты АН ( относительно воды как растворителя (
относительно воды как растворителя ( ) можно также записать в следующем виде:
) можно также записать в следующем виде:
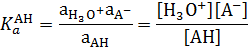
Размерность константы кислотности Ка моль·л–1. Значение Ка часто приводится в виде отрицательных десятичных логарифмов рКа (рКа = -lg Ka).
Стандартная константа кислотности кислоты Н3О+ равна:

Аналогично протону ион ОН– в растворах сольватирован: Н3О2–. Без учета сольватации ОН– взаимодействие основания В: с водой можно представить уравнением:

а выражение константы термодинамического равновесия реакции в следующем виде:
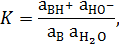
где  ‒ активности соответствующих частиц.
‒ активности соответствующих частиц.
Константа основности Кb основания В: в разбавленных водных растворах равна:


Амфотерные растворители SH (например, вода) склонны к автопротолизу:

который характеризуется константой автопротолиза растворителя SH:

Константы автопротолиза некоторых растворителей приведены в табл.14.1.
Таблица 14.1. Константы автопротолиза некоторых растворителей при 22 °С
| Растворитель
| Ион лиония
| Лиат‒ион
| р К авто= ‒lg К авто, моль2·л-2
|
| H2SO4
| H3SO4+
| HSO4-
| 3,33
|
| HCOOH
| HC(OH)2+
| HCO2-
| 6,2
|
| HF
| H2F+
| F-
| 12,5 (0°С)
|
| H2O
| H3O+
| HO-
|
|
| CH3COOH
| CH3COO+H2
| CH3COO-
| 14,45
|
| CH3OH
| CH3O+H2
| CH3O-
| 17,2
|
| CH3CH2OH
| CH3CH2O+H2
| CH3CH2O-
| 18,88
|
| (CH3)2C=O
| (CH3)2C=O+H
| CH2=C(CH3)O-
| 32,5
|
| CH3-C≡N
| CH3-C+=NH
| CH2=C=N-
| ≥33,3
|
Чем ниже константа автопротолиза растворителя, тем более сильные кислоты и основания могут существовать в этом растворителе.
Константа автопротолиза растворителя связана с константами кислотности кислоты и основности сопряженного основания соотношением:

Согласно этому соотношению, кислота тем сильнее, чем слабее сопряженное основание и наоборот. Для водных растворов при 22 °С:
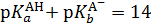
Кислоты условно делят на очень сильные ( р Ка < 0), сильные (0 < р Ка < 4,5), средней силы (4,5 < р Ка < 9), слабые (9 < р Ка < 14), очень слабые ( р Ка > > 14).
Для суперкислот (сверхкислот) в неводных средах р Ка < ‒10. К ним относятся 100 %-я НСlО4, безводная фторсульфоновая кислота FSO3H, смесь HF и SbF5, магическая кислота - эквимолярная смесь SbF6 и FSO3H, олеум различной концентрации.
Амфотерные растворители, такие, как Н2О, ROH, NH3, RCOOH, не только ограничивают область возможных значений р Ка , нооказывают сильное влияние на ионизационное равновесие. Например, сильные кислоты, растворенные в уксусной кислоте, хотя они не целиком ионизованы, образуют сольватированный протон СН3СОО+Н2, обладающий большей силой кислоты, чем Н3О+. Кислотность очень сильных кислот в воде делается идентичной силе кислоты сольватированного протона Н3О+ ( р Ка = ‒1,74), т. е. наблюдается выравнивающий эффект растворителя. Выравнивающим эффектом обладают "нивилирующие" растворители, в которых происходит полное превращение кислот в ониевые соли. Чем сильнее основные свойства растворителя, тем сильнее нивилирующее действие растворителя. Напротив, "дифференцирующими" являются малоосновные растворители, в которых проявляются индивидуальные свойства кислот (например, хлористый водород в бензоле).
14.2.2. Кислотность и основность реакционной среды. Функции кислотности
В химической технологии органического синтеза большинство процессов проводится в концентрированных растворах сильных минеральных кислот или оснований в воде, в полярных органических растворителях или в смесях воды с органическими растворителями. В этих случаях значения р Ка и р Кb не отражают в полной мере кислотные или основные свойства реакционной среды, так как ониотносятся к разбавленным растворам кислот и оснований в растворителях с высокой диэлектрической проницаемостью. Протонная теория кислот и оснований Брёнстеда предусматривает для каждого нового растворителя свою шкалу величин констант кислотности или основности, что вызывает большие неудобства.
Для создания абсолютной шкалы кислотности, позволяющей независимо от растворителя и концентрации кислоты (основания) оценивать протонирующую способность среды, Л. Гаммет и А. Дейруп (1932 г.) предложили применять индикаторный метод. В качестве стандарта они брали кислотность ионов гидроксония Н3О+ или гидроксила НО‒ в бесконечно разбавленных водных растворах. У индикаторов нейтральная (В:) и протонированная (ВН+) формы обладают разной цветностью, поэтому спектрофотометрическим методом легко определить концентрации этих форм. Имея набор индикаторов с разными константами кислотности и основности, измеренными в разбавленных водных растворах, можно определять абсолютную кислотность и основность любой среды.
При введении индикатора в количестве, не нарушающем протолитическое равновесие в реакционной системе, происходит протолитическая реакция между индикатором (В:) и средой. Для равновесия ионизации кислоты, сопряженной индикатору

константа кислотности индикатора в растворе равна:
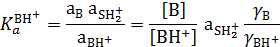
где  ‒ коэффициенты активности соответствующих частиц.
‒ коэффициенты активности соответствующих частиц.
Величина, связывающая активность протона, отдаваемого средой, с коэффициентами активности индикатора была названа Гамметом кислотностью среды (h 0):

На уравнении (14.7) основано определение кислотности среды - путем анализа цветности раствора индикатора с известной кислотностью его протонированной формы К аВН+.
Для разбавленных водных растворов  ,
,  принимается равной концентраций ионов гидроксония Н3О+ (как стандарту в шкале кислотности), следовательно h 0= [H3O+].
принимается равной концентраций ионов гидроксония Н3О+ (как стандарту в шкале кислотности), следовательно h 0= [H3O+].
По аналогии с разбавленными водными растворами пользуются и обратным логарифмов кислотности, который получил название функции кислотности Гаммета:

Очевидно, что эта функция аналогична рН = ‒lg[H3O+].
Кислотность среды h 0будет являться кислотной характеристикой раствора только в том случае, если отношение коэффициентов активности индикатора равно отношению коэффициентов активности какой-либо группы протонированных оснований (RН+) раствора, т. е.

Имея набор структурно-подобных индикаторов, в качестве которых Гаммет использовал замещенные нитроанилины, можно определить кислотность среды в широком диапазоне ее изменения по формуле:

За стандартное состояние для шкалы кислотности берут бесконечно разбавленный водный раствор сильной кислоты, где , а активность протонов равной их концентрации 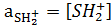 . В таких растворах
. В таких растворах
 .
.
Уравнение (14.10) принимает вид:

В точке "обесцвечивания" индикатора при [ВН+] = [В], получаем р Ка = рН. Следовательно, в разбавленных растворах можно определить значения р Ка индикаторов путем измерения значения рН растворов в точке обесцвечивания индикаторов.
Индикаторный метод измерения функции кислотности H0 состоит в следующем. Сначала выбирают наиболее сильное основание В, которое практически полностью протонируется в очень разбавленных растворах кислоты НА. Измерив отношение [B]/[BH+] и рН определяют  :
:
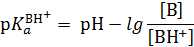
Зная р Ка и lg ([B]/[BH+]), находят значение Н 0винтервале рН индикатора. Это значение Н 0используется как начальный член шкалы Н 0.
Для определения функций кислотности более концентрированных растворов выбирают следующий индикатор D таким образом, чтобы, являясь более слабым основанием, он начинал протонироваться в тех растворах кислоты, где первый индикатор В еще не полностью протонирован. В этом "интервале перекрывания" определяют константу кислотности второго индикатора D:

Далее, измеряя  в более концентрированных растворах кислоты и зная
в более концентрированных растворах кислоты и зная  , определяют H0 для этих растворов. Затем аналогичную процедуру проводят со следующим индикатором и т. д. Таким образом, для определения H0 необходима серия индикаторов. Так, для системы HCl - H2O (до 16 M HCl) нужно 7-8 индикаторов, для водных р-ров H2SO4 от разбавленных до 100% H2SO4 - 10-12 индикаторов.
, определяют H0 для этих растворов. Затем аналогичную процедуру проводят со следующим индикатором и т. д. Таким образом, для определения H0 необходима серия индикаторов. Так, для системы HCl - H2O (до 16 M HCl) нужно 7-8 индикаторов, для водных р-ров H2SO4 от разбавленных до 100% H2SO4 - 10-12 индикаторов.
Численные значения функций кислотности, связанные с силой кислоты, представлены в табл. 14.2.
Таблица 14.2. Функции кислотности кислот
| Наименование кислоты
| Концентрация
| Функция кислотности
|
| Серная H2SO4
| 100 %
| Минус 11,1
|
| Серная H2SO4
| 10 моль/л
| Минус 4,89
|
| Перхлоратная HClO4
| 10 моль/л
| Минус 5,75
|
| Орто фосфорная H3PO4
| 10 моль/л
| Минус 2,59
|
Функция Н 0характеризует протонирующую силу кислоты в рассматриваемой среде, т. е. долю основания (индикатора или любого другого соединения с подобной структурой реакционного центра), переводимого в сопряженную кислоту.
Если теперь в среду с известной кислотностью вводить какой-то реагент R, то устанавливается протолитическое равновесие

и константа кислотности реагента определится выражением

Подставляя вместо  его выражение из уравнения (14.7), получим:
его выражение из уравнения (14.7), получим:

Отношение коэффициентов активности для одинаково заряженных форм сопряженных кислот и оснований является величиной постоянной (уравнение 14.9), что позволяет приравнять их сомножитель к единице и получить:

Уравнение (14.11) позволяет по известной кислотности данной среды (h0) и константе кислотности реагента рассчитать его протонированную форму. Следовательно, функции кислотности позволяют количественно описать кинетику кислотно-основного катализа в реальных растворах и сравнить результаты, полученные в разных растворителях.
Необходимо отметить, что метод Гаммета определения шкалы кислотности имеет свое ограничение. Уравнение (14.11) окажется неверным, если индикатор и реагент имеют разную зарядность исходной и протонированной формы. Если не соблюдается постулат Гаммета:

то функция кислотности Н 0не будет являться универсальной характеристикой протонирующей способности среды
Другую шкалу кислотности дает, в частности, функция НR, построенная на серии индикаторов, состоящих из замещенных арилкарбинолов ROH [C6lH5(CH3)2COH]:

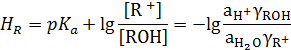
Для количественной характеристики основной среды отнимать протон у кислоты‒реагента пользуются функцией кислотности Н_, построенной с помощью индикаторов ВН, представляющих собой замещенные С‒Н‒кислоты общей формулы Аr СН2СН(СN)СН3:

Шкала Н– является продолжением шкалы рН в сторону повышенной основности.
Имеются также и другие шкалы кислотности, с которыми можно ознакомиться в специальной литературе по кислотно-основному равновесию.
14.2.3. Кинетика кислотно-основного катализа
Кислотный катализ протонными кислотами связан с активизацией реагента кислотой при протолитическом взаимодействии и последующем превращении активизированной частицы по моно- или бимолекулярной реакции в продукты превращения. К активизации протонными кислотами склонны реагенты-основания, имеющие свободные или лабильные электронные пары.
В водных растворах постоянное значение рН реакционной массы поддерживают обычно с помощью буферных растворов Они содержат слабую кислоту НА и сопряженное с ней основание А-, например, СН3СООН и СН3СОО-. В таких системах осуществляется равновесие:

Из уравнения кислотности кислоты НА значение рН раствора:
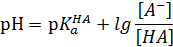
Это значение рН остается практически постоянным, так как при добавлении небольших количеств сильных кислот или оснований ионы Н3О+ или А-связываются основанием (кислотой) с образованием сопряженной кислоты (основания).
По формальному кинетическому признаку, характеризующему чувствительность скорости реакции к изменению общей концентрации буферного раствора кислотно-основный катализ подразделяется на специфический и общий.
 Рис. 14.2. Кинетический тест на наличие специфического и общего кислотного катализа: 1- специфический катализ, 2 – общий катализ
Рис. 14.2. Кинетический тест на наличие специфического и общего кислотного катализа: 1- специфический катализ, 2 – общий катализ
| Типичный вид зависимости скорости таких реакций от рН среды представлен на рис. 14.2.
К специфическому катализу относится катализ, при котором скорость реакции определяется только значением рН или концентрацией ионов гидроксония и не зависит от концентрации других кислот и оснований, находящихся в растворе.
К общему катализу относится тип катализа, при котором скорость катализа зависит от рН и от концентрации других кислот и оснований, находящихся в растворе
|
14.2.3.1. Специфический кислотный катализ
Специфический кислотный катализимеет место, если каталитический эффект вызван присутствием протона в активированном комплексе. Это связано с быстрым установлением всех протолитических равновесий в растворе и последующем медленном превращении протонированного реагента в продукты реакции.

Одно из этих уравнений стехиометрически зависимо от других и для количественной характеристики материального баланса можно ограничиться двумя последними.
Последующие превращения RH+ на лимитирующей стадии могут протекать мономолекулярно (механизм А-1)

Либо с участием второго исходного реагента Y (механизм А-2):

Протонированный продукт реакции РН+ и второй исходный Y, которой всегда является нуклеофилом и обладает основными свойствами, также участвуют в быстрых протолитических реакциях

Скорость реакции, протекающей по механизмам А-1, описывается кинетическим уравнением:

Для практического использования необходимо преобразовать эти уравнения в форму, в которой действительные концентрации выражены аналитическими (суммарными) концентрациями и через известную концентрацию иона лиония или h 0.
Степень протонирования реагента [RH+]/[R] однозначно определяется кислотностью среды h 0 по уравнению (14.11).
Кинетические уравнения имеют наиболее простой вид при постоянстве h 0 в ходе реакции. Это условие выполняется:
1. при близкой или малой основности продукта Р и исходных реагентов R и Y, так что кислотность среды не изменяется в ходе реакции;
2. при проведении реакции в буферном растворе;
3. в концентрированном растворе кислоты АН, взятой в избытке по отношению к исходным реагентам R и Y.
Аналитическая (суммарная) концентрация реагента R с учетом уравнения 14.11 равна:

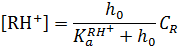
Тогда выражение скорости реакции можно записать в следующем виде:


Прологарифмируем уравнение (14.14)

График зависимости (14.15) представлен на рис. 14.3, а.
 а
а
|  б
б
|
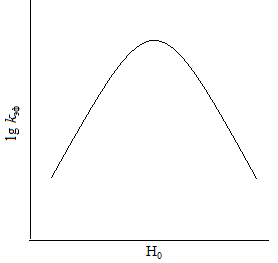
| Рис. 14.3. Зависимость lg k эф от функции кислотности среды Н0 для реакций кислотно-основного катализа
|
При малой кислотности и слабых основных свойств реагентов  уравнение (14.15) принимает вид
уравнение (14.15) принимает вид

что соответствует правому нисходящему линейному участку кривой.
При высокой кислотности среды, когда  , в уравнении (14.15) можно пренебречь константой кислотности и экспериментально определяемая эффективная константа скорости не зависит от кислотности среды и равна истинной константе скорости k эф= k 2. Это указывает на то, что в процессе активизации произошло количественное протонирование реагента и дальнейшее повышение кислотности среды не приводит к увеличению концентрации [RH+] и k ЭФ (горизонтальный участок кривой).
, в уравнении (14.15) можно пренебречь константой кислотности и экспериментально определяемая эффективная константа скорости не зависит от кислотности среды и равна истинной константе скорости k эф= k 2. Это указывает на то, что в процессе активизации произошло количественное протонирование реагента и дальнейшее повышение кислотности среды не приводит к увеличению концентрации [RH+] и k ЭФ (горизонтальный участок кривой).
Криволинейный участок этой зависимости отвечает соизмеримым значениям  и h 0 и описывается уравнением (14.15).
и h 0 и описывается уравнением (14.15).
Уравнением (14.13) описываются реакции третичных спиртов и их эфиров, реакции гидролиза и алкоголиза уксусного ангидрида и т. п. Эти реакции ускоряются небольшими добавками сильных кислот, скорость реакций пропорциональна кислотности (h 0, [Н3О+]) и не зависит ни от концентрации, ни от природы нуклеофильного реагента.
При протекании реакции по механизму А-2 скорость реакции описывается уравнением:

которое включают концентрацию протонированной формы реагента и свободной формы нуклеофила Y.
Преобразование кинетического уравнения реакции, протекающей по механизму А-2, путем выражения концентраций RH+ и свободного реагента Y через суммарные концентрации этих реагентов дает:



Из уравнения (14.17) следует, что при h0 = const зависимость концентраций реагентов от времени для реакции, протекающей по механизму А-2, описывается кинетическим уравнением второго порядка, в котором экспериментально определяемая константа скорости зависит от кислотности

Из уравнения (4.18) видно, что зависимость  от h 0 должна проходить через максимум. Зависимость (14.18) в логарифмических координатах дает кривую (б) на рис. 14.3. Для нахождения координат максимума h 0 продифференцируем по h0 и полученное выражение приравняем к нулю. Таким образом, найдем, что максимальное значение соответствует hmax равно:
от h 0 должна проходить через максимум. Зависимость (14.18) в логарифмических координатах дает кривую (б) на рис. 14.3. Для нахождения координат максимума h 0 продифференцируем по h0 и полученное выражение приравняем к нулю. Таким образом, найдем, что максимальное значение соответствует hmax равно:

При кислотности среды меньше  , когда
, когда 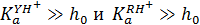 ,выражение эффективной константы скорости принимает вид
,выражение эффективной константы скорости принимает вид

и ему соответствует правая нисходящая ветвь кривой (б) рис. 14.3. В этих условиях степень протонирования как R, так и Y незначительна, суммарная концентрация CY практически равна концентрации свободного реагента Y, a [RH+] возрастает прямо пропорционально h 0.
При высоких значениях кислотности среды (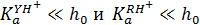 ) выражение для эффективной константы примет вид:
) выражение для эффективной константы примет вид:

Из полученного выражения следует, что при высокой кислотности среды реагенты R и Y практически полностью протонированы и снижение скорости реакции связано с уменьшением незначительной доли еще не протонированного реагента Y, концентрация которого обратно пропорциональна кислотности среды.
Наличие максимума скорости характерно для многих реакций, протекающих по механизму А-2 в концентрированных растворах кислот. К ним относятся, в частности, реакции нитрования ароматических соединений.
Такие же экстремальные зависимости характерны для гидролиза органических амидов, катализируемого кислотами:
 <
<