В результате липолиза в крови увеличивается содержание жирных кислот, а в печени усиливается синтез липопротеидов. Но печень вследствие недостаточного синтеза апопротеина не в состоянии полностью усвоить поступающие жирные кислоты, идущие на построение липопротеидов. Избыток жирных кислот синтезируется в печени в триглицериды. При сахарном диабете, сопровождающемся общим ожирением (что отмечается довольно часто), стеатоз печени усиливается в связи с избыточным поступлением жиров и углеводов с пищей. При этом основным механизмом поступления жира в печень остается липолиз, ведущий к гиперлипидемии.
Морфологической особенностью жировой дистрофии печени при сахарном диабетеявляется вакуолизация ядер ожиревших гепатоцитов за счет накопления в них гликогена — "дырчатые", или "гликогенные", ядра. Жир в цитоплазме гепатоцитов (стеатоз) и "дырчатые" их ядра (накопление гликогена) — характерные гистологические признаки диабетической жировой дистрофии печени.
Интоксикации. Жировая дистрофия печени развивается при воздействии на организм таких токсичных веществ, как четыреххлористый углерод, гидразин-сульфат, тетрахлорэтан, ДДТ, фосфор, а также ряда лекарственных средств тетрациклины, стероиды, барбитураты, метотрексат и др.).
В этих условиях накопление липидов в гепатоцитах обусловлено, как правило, нарушением синтеза белка (апопротеина) вследствие блокады их ферментных систем. Недостаток апопротеина вызывает нарушение синтеза липопротеидов, способных проникать через наружную мембрану гепатоцитов. Задержка липидов в цитозоле приводит к образованию триглицеридов. Накопление жира в гепатоцитах связано и с распадом липопротеидных комплексов мембран гепатоцитов, т.е. с механизмом фанероза.
Миокард. Развитие жировой дистрофии миокарда (рис. 9) связывают с тремя основными механизмами:

Рис. 9. Жировая дистрофия миокарда (окраска Суданом III).
В цитоплазме мышечных клеток, расположенных преимущественно вокруг венул и вен, видны скопления мелких капель жира желто-красного цвета (1); другие мышечные клетки свободны от жировых включений (2). Отсутствует поперечная исчерченность мышечных клеток, ядро сморщено или лизировано
1) повышенным поступлением жирных кислот в кардиомиоциты;
2) нарушением обмена жиров в этих клетках;
3) распадом липопротеидных комплексов внутриклеточных структур, т.е. фанерозом.
Основой этих трех механизмов жировой дистрофии кардиомиоцитов является энергетический дефицит миокарда.
Известно, что в кардиомиоциты липиды поступают в виде жирных кислот, освобождающихся с помощью липопротеидлипазы от плазменных триглицеридов или связей с альбуминами. Жирные кислоты используются миокардом для энергетических нужд (что достигается их (окислением в митохондриях) и для построения структурных фосфолипидов. Из этого следует, что при любых состояниях, сопровождающихся энергетическим дефицитом, усиливается поступление в миокард жирных кислот, из которых синтезируются нейтральные жиры. Механизм фанероза в развитии жировой дистрофии кардиомиоцитов заключается не в высвобождении липидов из липопротеидных комплексов мембранных структур клетки, а в нарушении окисления поступающих в избытке в клетку жирных кислот при деструкции ее митохондрий.
Причины развития жировой дистрофии миокарда следующие:
1) гипоксия (при анемиях, хронической сердечно-сосудистой недостаточности);
2) интоксикации (дифтерийная, алкогольная, отравление фосфором, мышьяком, хлороформом и др.)
Гипоксия. Это наиболее частая причина жировой дистрофии миокарда, поскольку гипоксия ведет к энергетическому дефициту высокоспециализированных тканей, к которым относится миокард. Недостаток кислорода нарушает процессы окислительного фосфорилирования в кардиомиоцитах, что приводит к переключению обмена миокарда на анаэробный гликолиз и резкому снижению количества АТФ. Дефицит энергии усиливается в связи с нарастающим ацидозом ткани; развивается повреждение митохондрий, нарушается окисление жирных кислот, и липиды накапливаются в кардиомиоцитах чаще в виде мелких капель (различают также и пылевидное ожирение миокарда).
Жировая дистрофия миокарда чаще имеет очаговый характер — содержащие жир кардиомиоциты расположены преимущественно по ходу венозного колена капилляров и мелких вен, где гипоксический фактор наиболее резко выражен. Очаговостью поражения объясняется своеобразный внешний вид сердца: со стороны эндокарда, особенно в области сосочковых мышц, видна желтовато-белая исчерченность ("тигровое сердце"); миокард дряблый, бледно-желтый, камеры сердца растянуты, размеры его несколько увеличены.
Интоксикации. Наиболее изучена жировая дистрофия миокарда при дифтерийной и алкогольной интоксикации.
При дифтерийной интоксикации накопление липидов в кардиомиоцитах обусловлено снижением интенсивности их окисления вследствие как недостатка карнитина, так и повреждения митохондрий.При алкогольной интоксикации также имеют место снижение интенсивности окисления жирных кислот в кардиомиоцитах и деструкция их митохондрий, что ведет к резкому уменьшению активности ферментов.
Морфологические изменения сердца при интоксикации подобны таковым при гипоксии, но при дифтерии по сравнению с алкоголизмом они выражены сильнее.
Почки. Следует помнить, что нейтральные жиры обнаруживаются в эпителии узкого сегмента и собирательных трубочек и в физиологических условиях. О жировой дистрофии почек говорят в тех случаях, когда липиды (нейтральные жиры, холестерин, фосфолипиды) появляются в эпителии канальцев главных отделов нефрона — проксимальных и дистальных.
Наиболее часто жировая дистрофия почек встречается при нефротическом синдроме и хронической почечной недостаточности, реже — при инфекциях и интоксикациях.
Нефротический синдром. Как уже упоминалось ранее, нефротический синдром характеризуется не только массивной протеинурией, обусловливающей развитие отеков и гипо-, диспротеинемии, но и гиперлипидемией, повышением в крови уровня триглицеридов, холестерина и фосфолипидов. Гиперлипидемию в этих случаях объясняют увеличением синтеза холестерина и мобилизацией жира из жировых депо, снижением активности липопротеидлипазы и холестеринлецитинацетилтрансферазы в сыворотке крови, усилением синтеза липидов в почках вследствие угнетения почечной липолитической активности.
Понятно, что гиперлипидемия обусловливает липидурию, главным образом за счет липопротеидов. В условиях характерной для нефротического синдрома повышенной проницаемости гломерулярного фильтра липиды подвергаются повышенной резорбции эпителием канальцев, загружая не только цитоплазму нефроцитов, но и строму почки.
Жировая дистрофия нефроцитов при нефротическом синдроме присоединяется к гиалиново-капельной и гидропической, о которых уже шла речь ранее.
Хроническая почечная недостаточность. При этом синдроме уровень триглицеридов и холестерина в крови также повышен. Это связывают со снижением активности липопротеидлипазы и уменьшением утилизации глюкозы, что приводит к усилению липолиза. Снижение утилизации глюкозы обусловлено дефицитом белка в пищевом рационе больных с хронической почечной недостаточностью (уремией). Дефицит белка подавляет синтез ферментов, необходимых для процессов окисления.
Морфологические изменения почек при жировой их дистрофии достаточно характерны. При микроскопическом исследовании липиды видны в цитоплазме эпителия канальцев и строме почки в виде капель (нейтральный жир) или двояко-преломляющих кристаллов (холестерин). Почки при нефротическом синдроме увеличены, дряблые, с желтым крапом на поверхности (при амилоидозе почек, сопровождающемся нефротическим синдромом, они плотные, с сальным блеском на разрезе). При хронической почечной недостаточности почки уменьшены, чаще с зернистой поверхностью, серо-желтые, с истонченным корковым веществом.
Группу наследственных липидозов составляют так называемые системные липидозы, возникающие вследствие наследственного дефицита ферментов, участвующих в метаболизме определенных липидов. Поэтому системные липидозы относят к наследственным ферментопатиям (болезни накопления), поскольку дефицит фермента определяет накопление субстрата, т. е. липидов, в клетках.
В зависимости от вида накапливающихся в клетках липидов различают: цереброзидлипидоз, или глюкозилцерамидлипидоз (болезнь Гоше), сфингомиелинлипидо з (болезнь Ниманна—Пика), ганглиозидлипидоз (болезнь Тея— Сакса, или амавротическая идиотия), генерализованный ганглиозидоз (болезнь Нормана—Ландинга) и др. Чаще всего липиды накапливаются в печени, селезенке, костном мозге, центральной нервной системе (ЦНС), нервных сплетениях. При этом появляются характерные для того или иного вида липидоза клетки (клетки Гоше, клетки Пика), что имеет диагностическое значение при изучении биоптатов. Многие ферменты, дефицит которых определяет развитие системных липидозов, относятся, к лизосомным. На этом основании ряд липидозов рассматривают как лизосомные болезни..
УГЛЕВОДНЫЕ ДИСТРОФИИ.
Углеводы, которые определяются в клетках и тканях и могут быть идентифицированы гистохимически, делят на полисахариды, из которых в животных тканях выявляются лишь гликоген, гликозаминогликаны (мукополисахариды) и гликопротеиды.
Среди гликозаминогликанов различают нейтральные, прочно связанные с белками, и кислые, к которым относятся гиалуроновая, хондроитинсерная кислоты и гепарин. Кислые гликозаминогликаны как биополимеры способны вступать в непрочные соединения с рядом метаболитов и осуществлять их транспорт. Главными представителями гликопротеидов являются муцины.
2.3.1. Наследственные углеводные дистрофии, в основе которых лежат нарушения обмена гликогена, называются гликогенозами. Гликогенозы обусловлены отсутствием или недостаточностью фермента, участвующего в расщеплении депонированного гликогена, и относятся поэтому к наследственным ферментопатиям, или болезням накопления.
В настоящее время хорошо изучены 6 типов гликогенозов, обусловленных наследственной недостаточностью 6 различных ферментов. Это болезни Гирке (I тип), Помпе (II тип), Мак-Ардля (V тип) и Герса (VI тип), при которых структура накапливаемого в тканях гликогена не нарушена, и болезни Форбса—Кори (III тип) и Андерсена (IV тип), при которых она резко изменена.
Морфологическая диагностика гликогеноза того или иного типа возможна при биопсии с помощью гистоферментативных методов.
2.3.2. Углеводные дистрофии, связанные с нарушением обмена гликопротеидов. При нарушении обмена гликопротеидов в клетках или в межклеточном веществе происходит накопление муцинов и мукоидов, называемых также слизистыми или слизеподобными веществами. В связи с этим при нарушении обмена гликопротеидов говорят о слизистой дистрофии.
Микроскопическое исследование. Оно позволяет выявить не только усиленное слизеобразование, но и изменения физико-химических свойств слизи. Многие секретирующие клетки погибают и десквамируются, выводные протоки желез обтурируются слизью, что ведет к развитию кист. Нередко в этих случаях присоединяется воспаление. Слизь может закрывать просветы бронхов (рис. 10), следствием чего является возникновение ателектазов и очагов пневмонии.
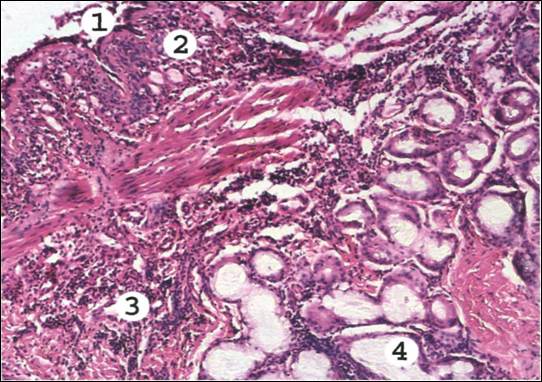
Рис. 10. Биоптат бронха при бронхиальной астме.
В просвете бронха слоистый слизистый секрет с примесью эозинофилов и десквамированного эпителия (1). Слизистая оболочка инфильтрирована эозинофилами, базофилами, лимфоидными и плазматическими клетками (2). Базальная мембрана эпителия утолщена, набухшая. Сосуды микроциркуляторного русла резко полнокровны, их проницаемость повышена, отмечается серозный периваскулярный отек слизистой оболочки и подслизистого слоя (3). Слизистые железы стенки бронха в состоянии гиперсекреции, их протоки расширены, заполнены слизью (4). Мышечная оболочка гипертрофирована.
Причиныслизистой дистрофии разнообразны, но чаще всего это воспаление слизистых оболочек в результате действия различных патогенных раздражителей.
Муцины составляют основу слизи, продуцируемой эпителием слизистых оболочек и железами, мукоиды входят в состав многих тканей.
Иногда в железистых структурах накапливается не истинная слизь, а слизеподобные вещества (псевдомуцины). Эти вещества могут уплотняться и принимать характер коллоида. Тогда говорят о коллоидной дистрофии, которая наблюдается, например, при коллоидном зобе (рис. 11).
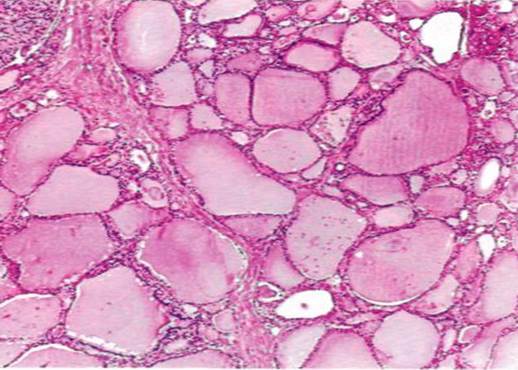
Рис. 11. Коллоидный зоб