

Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Биохимия спиртового брожения: Основу технологии получения пива составляет спиртовое брожение, - при котором сахар превращается...
Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Биохимия спиртового брожения: Основу технологии получения пива составляет спиртовое брожение, - при котором сахар превращается...
Топ:
Марксистская теория происхождения государства: По мнению Маркса и Энгельса, в основе развития общества, происходящих в нем изменений лежит...
Когда производится ограждение поезда, остановившегося на перегоне: Во всех случаях немедленно должно быть ограждено место препятствия для движения поездов на смежном пути двухпутного...
Характеристика АТП и сварочно-жестяницкого участка: Транспорт в настоящее время является одной из важнейших отраслей народного...
Интересное:
Уполаживание и террасирование склонов: Если глубина оврага более 5 м необходимо устройство берм. Варианты использования оврагов для градостроительных целей...
Искусственное повышение поверхности территории: Варианты искусственного повышения поверхности территории необходимо выбирать на основе анализа следующих характеристик защищаемой территории...
Аура как энергетическое поле: многослойную ауру человека можно представить себе подобным...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Перман-ски в щелочной среде, определяют орг соед-я
Признаком окончания титрования служит бледно-розовая окраска избытка титранта. Поэтому, если титруемый раствор бесцветен, о достижении ТЭ можно судить по появлению бледно-розовой окраски избытка титранта КМnО4 при титровании прямым способом или по исчезновению окраски при обратном титровании.
ПЛЮСЫ 1) возможность титрования раствором КМnО4 в любой среде (кислой, нейтральной, щелочной);
2) применимость растворов перманганата калия в кислой среде для определения многих веществ, которые не взаимодействуют с более слабыми окислителями; 3) стехиометричность и достаточно высокую скорость большинства окислительно-восстановительных реакций с участием МnО4- при оптимально выбранных условиях;
4) возможность титрования без индикатора;
5) доступность перманганата калия.
МИНУСЫ 1) титрант КМnО4 готовят как вторичный стандарт, тк трудно получить в чистом состоянии;
2) реакции с участием МnО4- возможны в определенных условиях (рН, температура);
3) титрование раствором КМnО4 не рекомендуется проводить в присутствии С1-, что затрудняет определение некоторых веществ.
Определение восстановителей. Если ОВР между определяемым восстановителем и МnО4- протекает быстро, то титрование проводят прямым способом. Так определяют оксалаты, нитриты, пероксид водорода, железо
Например:
5Н2О2 + 2МnО4- + 6Н+ → 5О2 + 2Мn2+ + 8Н2О,
5[Fe(CN)6]4- + МnО4- + 8Н+ → 5[Fe(CN)6]3- + Мn2+ + 4Н2О,
В случае замедленных реакций определение проводят способом обратного титрования избытка перманганата.
Так определяют муравьиную, поли- и оксикарбоновые кислоты, альдегиды и другие органические соединения, например:
НСОО- + 2MnO4- + 3OH- → CO32- + 2MnO42- + 2Н2O
|
|
Затем избыток перманганата оттитровывают щавелевой кислотой или оксалатами:
2MnO4- + 5C2O42- + 16H+ → 2Mn2+ + 10CO2 + 8H2O
Определение окислителей. Добавляют избыток стандартного раствора восстановителя и затем титруют его остаток раствором KMnO4 (способ обратного титрования). Например, дихроматы, персульфаты, хлориты и другие окислители можно определять перманганатометрическим методом, подействовав сначала избытком стандартного раствора Fe2+, а затем оттитровав непрореагировавшее количество Fe2+ раствором KMnO4:
Cr2O72- + 6Fe2+ + 14H+ → 2Cr3+ + 6Fe3+ + 7H 2O
Титрование избытка ионов Fe2+ проводят перманганатом (вспомогательным рабочим раствором):
5Fe2+ + MnO-4 + 8H+ → 5Fe3+ + Mn2+ + 4H2O.
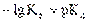 Определение веществ, не обладающих окислительно-восстановительными свойствами, проводят косвенным способом, например титрованием по замещению. Для этого определяемый компонент переводят в форму соединения, обладающего восстановительными или окислительными свойствами, а затем проводят титрование. Например, ионы кальция, цинка, кадмия, кобальта осаждают в виде малорастворимых оксалатов
Определение веществ, не обладающих окислительно-восстановительными свойствами, проводят косвенным способом, например титрованием по замещению. Для этого определяемый компонент переводят в форму соединения, обладающего восстановительными или окислительными свойствами, а затем проводят титрование. Например, ионы кальция, цинка, кадмия, кобальта осаждают в виде малорастворимых оксалатов
М2+ + С2О42- → МС2О4.
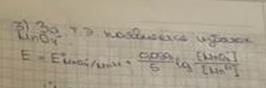
 Определение органических соединений. Отличительной особенностью реакций органических соединений с MnO4- является их малая скорость. Определение возможно, если использовать обратное титрование: анализируемое соединение предварительно обрабатывают избытком сильнощелочного раствора перманганата и дают возможность реакции протекать необходимый период времени. Остаток перманганата титруют раствором оксалата натрия. Например, при определении глицерина протекают реакции:
Определение органических соединений. Отличительной особенностью реакций органических соединений с MnO4- является их малая скорость. Определение возможно, если использовать обратное титрование: анализируемое соединение предварительно обрабатывают избытком сильнощелочного раствора перманганата и дают возможность реакции протекать необходимый период времени. Остаток перманганата титруют раствором оксалата натрия. Например, при определении глицерина протекают реакции:
C3H5(OH)3 + 14MnO4- +20 OH- → 3CO32-+ 14MnO42- + 14H2O,
2MnO4- +5C2O42- + 16H+ → 2Mn2++ 10CO2 + 8H2O.
Кривая титрования перманганотометрии
!!!!!! Буферные растворы применяют в химическом анализе в тех случаях, когда по условиям опыта химическая реакция должна протекать при соблюдении точного значения pH, не меняющегося при разбавлении раствора или при добавлении к нему других реагентов. Например, при проведении реакций окисления—восстановления, при осаждении сульфидов, гидроокисей, карбонатов, хроматов, фосфатов и др.
|
|
Каждая из буферных систем характеризуется определенной присущей ей концентрацией ионов Н+ (активной кислотностью), которую система и стремится сохранить на неизменном уровне при добавлении к ней сильной кислоты либо щелочи.
Установим на примере ацетатного буфера факторы, влияющие на величину активной кислотности.
В растворе данной буферной системы происходят следующие реакции электролитической диссоциации:
CH3COOH →CH3COO– + H+
CH3COONa → CH3COO– + Na+
(Гидролиз соли, т.е. взаимодействие ацетат-ионов с Н2О: CH3COO– + HOH → CH3COOH + OH– учитывать не будем.)
Таким образом, ионы Н+ образуются только за счет диссоциации некоторого числа молекул уксусной кислоты. Этот процесс является обратимым и количественно характеризуется константой кислотности Kа:
 Где CH+ (или CH3O+), СCH3COO- и СCH3COOH - равновесные молярные концентрации ионов Н+, СН3СОО– и непродиссоциированных молекул кислоты.
Где CH+ (или CH3O+), СCH3COO- и СCH3COOH - равновесные молярные концентрации ионов Н+, СН3СОО– и непродиссоциированных молекул кислоты.
Из данного уравнения можно выразить CH+ (активную кислотность буферной системы):
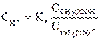 Кроме уксусной кислоты, в растворе присутствует ее соль CH3COONa. Она является сильным электролитом и полностью распадается на ионы. В результате этого концентрация анионов СН3СОО– резко возрастает, и согласно принципу Ле-Шателье, равновесие реакции диссоциации уксусной кислоты смещается влево, т.е. в сторону образования ее молекул. Причем диссоциация уксусной кислоты в присутствии собственной соли может быть настолько подавленной, что равновесную концентрацию ее нераспавшихся молекул в растворе можно считать равной концентрации СН3СООН, а равновесную концентрацию ацетат-ионов – исходной концентрации соли. В связи с этим выражение, по которому рассчитывается концентрация ионов Н+, можно записать иначе:
Кроме уксусной кислоты, в растворе присутствует ее соль CH3COONa. Она является сильным электролитом и полностью распадается на ионы. В результате этого концентрация анионов СН3СОО– резко возрастает, и согласно принципу Ле-Шателье, равновесие реакции диссоциации уксусной кислоты смещается влево, т.е. в сторону образования ее молекул. Причем диссоциация уксусной кислоты в присутствии собственной соли может быть настолько подавленной, что равновесную концентрацию ее нераспавшихся молекул в растворе можно считать равной концентрации СН3СООН, а равновесную концентрацию ацетат-ионов – исходной концентрации соли. В связи с этим выражение, по которому рассчитывается концентрация ионов Н+, можно записать иначе:
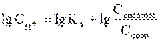
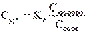 где Скислоты и Ссоли – исходные концентрации компонентов буферной системы.
где Скислоты и Ссоли – исходные концентрации компонентов буферной системы.

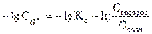 Прологарифмируем полученное уравнение (с учетом того, что логарифм произведения равен сумме логарифмов сомножителей):
Прологарифмируем полученное уравнение (с учетом того, что логарифм произведения равен сумме логарифмов сомножителей):
и умножим обе его части на -1:

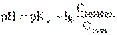 В связи с этим запишем уравнение для расчета концентрации ионов Н+ в окончательном виде:
В связи с этим запишем уравнение для расчета концентрации ионов Н+ в окончательном виде:
 Значение буферов в комплексонометрии:
Значение буферов в комплексонометрии:
1. Необходимо создать оптимальное значение рН. Нижняя граница оптимального для титрования значения рН определяется тем значением рН, при котором комплексонат еще достаточно устойчив. Верхняя граница этого интервала зависит от значения рН, при котором начинают образовываться гидроксокомплексы или осадок гидрокcида. Для предотвращения образования осадка в титруемый раствор иногда добавляют комплексообразующис вещества (аммиак, тартраты, и.т. п.). Обшая закономерность такова: чем больше заряд иона металла, тем более прочные комплексонаты он образует и тем более склонен к гидролизу, поэтому тем меньшим оказывается оптимальное для титрования значение рН.
2. Необходимо поддерживать определенное значение рН с помощью буферного раствора. Любое комплексонометрическое титрование проводят в присутствии буфера.
|
|
Прочность комплексной связи зависит от pH раствора. При сравнительно высоких значениях pH прочность комплексов различных элементов примерно одинакова, но при pH = 2,6 разница в прочности комплексных ионов наибольшая. Устойчивость комплексных соединений
 зависит от природы входящих в комплекс аддендов и реакции среды. Некоторые комплексные соединения устойчивы в широком диапазоне pH в кислой среде, другие — в щелочной, но имеются такие комплексы, которые могут существовать лишь в определенных, довольно узких интервалах значений pH. С марганцем, кобальтом, никелем, цинком, а также с медью и кадмием образуются растворимые комплексные соединения, устойчивые лишь при определенной величине pH раствора.
зависит от природы входящих в комплекс аддендов и реакции среды. Некоторые комплексные соединения устойчивы в широком диапазоне pH в кислой среде, другие — в щелочной, но имеются такие комплексы, которые могут существовать лишь в определенных, довольно узких интервалах значений pH. С марганцем, кобальтом, никелем, цинком, а также с медью и кадмием образуются растворимые комплексные соединения, устойчивые лишь при определенной величине pH раствора.

Билет №6
!!!!! Основной полуреакцией методов хроматометрии является процесс окисления дихроматом калия в кислой среде: Cr2O72- + 14H+ +6e- → 2Cr3+ + 7H2O;
По основному реактиву эту группу методов иногда называют дихроматометрией. Как показывает величина стандартного окислительно-восстановительного потенциала,дихромат является менее сильным окислителем, чем перманганат.
Рабочим раствором хроматометрии является раствор дихромата калия K2Cr2O7. Кристаллический препарат K2Cr2O7 легко приготовить достаточно чистым, он устойчив при хранении на воздухе, поэтому титрованные растворы дихромата калия готовят по точной навеске кристаллической соли. Раствор дихромата калия сохраняет неизменный титр длительное, неопределенно долгое время, его можно кипятить без разложения. Железо (II) титруется дихроматом в солянокислой среде без каких-либо побочных процессов и осложнений. Недостатком реактива является образование в результате реакции окрашенных ионов Cr3+, затрудняющих своей окраской фиксирование ТЭ. Поэтому часто возникает необходимость в применении индикаторов. В качестве индикаторов используют дифениламин, а также дифениламинсульфоновую кислоту, фенилантраниловую кислоту и др. Наиболее важными практическими применениями хроматометрии являются определение железа в различных пробах после предварительного восстановления его до Fe(II), а также урана, который предварительно переводят в U(IV). Кроме того, реакция титрования железа(II) дихроматом является заключительным этапом различных аналитических методик, основанных на реакциях взаимодействияFe(III) или Fe(II) с определяемым веществом. Восстановители анализируют по методу замещения. Например, Cu(I) реагирует с Fe(III) в кислой среде:
|
|
 Cu+ + Fe3+ = Fe2+ + Cu2+. В результате реакции в р-ре появ. к-во ионов Fe2+, эквивалентное количеству Cu(I) в исходной пробе. Окислители определяют методом обратного титрования. Например, хром в сталях окисляют до Cr2O72-, добавляют избыток титрованного раствора соли Мора (NH4)2Fe(SO4)2.6H2O, и не вошедшее в реакцию количество оттитровывают дихроматом.
Cu+ + Fe3+ = Fe2+ + Cu2+. В результате реакции в р-ре появ. к-во ионов Fe2+, эквивалентное количеству Cu(I) в исходной пробе. Окислители определяют методом обратного титрования. Например, хром в сталях окисляют до Cr2O72-, добавляют избыток титрованного раствора соли Мора (NH4)2Fe(SO4)2.6H2O, и не вошедшее в реакцию количество оттитровывают дихроматом.
Нитраты в растворе определяют, обрабатывая пробу раствором соли Мора при кипячении в инертной атмосфере в присутствии молибдата как катализатора:
3Fe2+ + NO-3 + 4H+ = 3Fe3+ + NO + 2H2O.
После охлаждения раствор титруют дихроматом.
Некоторые катионы титруют дихроматом непосредственно (Sn2+, Sb3+ и др.).
Точка эквивалентности в хроматометрии определяется с помощью небольшого количества дифениламина, при малейшем избытке дихромата окрашивающего раствор в синий цвет.
!!!!! 1. Молярная концентрация или молярность [С(В) или СВ] моль/л –показывает сколько моль вещества В содержится в 1 литре раствора. С(В)=m(г)/(Мр.в. ·Vр-ра)
2. Молярная концентрация эквивалента или нормальность [C(fэкв(B)B)] моль×экв/л -показывает сколько моль эквивалента вещества В содержится в 1 литре раствора.
C(fэкв(B)B)= m(г)/(Э ·Vр-ра)
Эквивалент вещества В [ЭВ]- частица этого вещества, эквивалентная одному иону водорода в реакциях КОВ
или одному электрону в реакциях окисления-восстановления.
Фактор эквивалентности [fэкв(В)] –безразмерная величина, равная или меньше единицы. Рассчитывается на основании стехиометрии реакции:
Молярная масса эквивалента вещества В:
[M(fэкв(B)B)]= fэкв(B)×M(B)
Массовая доля или процентная концентрация -отношение массы (г) данного вещества в растворе к массе всего раствора.
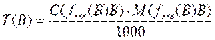 3. Титр рабочего раствора В по рабочему веществу В [T(B) или ТВ] г/мл –число граммов растворенного вещества В в 1 мл раствора
3. Титр рабочего раствора В по рабочему веществу В [T(B) или ТВ] г/мл –число граммов растворенного вещества В в 1 мл раствора
|
|
Т(В) = m/V, или через концентрацию
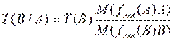 4. Титр рабочего раствора В по определяемому веществу А или условный титр [ТВ/А или Т(В/А)] г/мл -число граммов определяемого вещества А, эквивалентное 1 мл рабочего раствора В.
4. Титр рабочего раствора В по определяемому веществу А или условный титр [ТВ/А или Т(В/А)] г/мл -число граммов определяемого вещества А, эквивалентное 1 мл рабочего раствора В.

Обычно концентрацию раствора хорошо растворимых твердых или жидких веществ выражают весовым количеством данного вещества, содержащегося в определенном весовом количестве (или объеме) раствора или растворителя.
Существуют следующие способы выражения концентрации растворов.
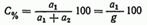 1. В процентах (С%), т. е. числом граммов растворенного вещества, находящегося в 100 г раствора:
1. В процентах (С%), т. е. числом граммов растворенного вещества, находящегося в 100 г раствора:
где a1 — количество растворенного вещества, г; a2— количество растворителя, г; g— масса раствора, равная а1+а2, г.

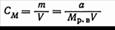 2. В единицах молярности, т. е. числом грамм-молекул растворенного вещества в 1л раствора: где а — количество растворенного вещества, г; m — число грамм-молекул растворенного вещества; V — объем раствора, л; Mрв— масса 1 моль растворенного вещества (численно равная молекулярному весу), г/моль.
2. В единицах молярности, т. е. числом грамм-молекул растворенного вещества в 1л раствора: где а — количество растворенного вещества, г; m — число грамм-молекул растворенного вещества; V — объем раствора, л; Mрв— масса 1 моль растворенного вещества (численно равная молекулярному весу), г/моль.
Если V=1, то формула (2) принимает вид: Очень часто См изображают просто индексом С. При числах молярность выражают буквой М.
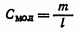 Иногда концентрацию выражают числом молей вещества не на 1000мл раствора, а на 1000 г растворителя. Такие растворы, в отличие от молярных, называют моляльными растворами:
Иногда концентрацию выражают числом молей вещества не на 1000мл раствора, а на 1000 г растворителя. Такие растворы, в отличие от молярных, называют моляльными растворами:
где m - число молей растворенного вещества; l — число тысяч граммов растворителя.
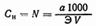
 3. В единицах нормальности, т. е. числом грамм-эквивалентов (Э) вещества, растворенного в 1л раствора: где а — количество растворенного вещества, г; n — число грамм-эквивалентов; Если навеска исходного вещества (а) растворена в V миллилитрах, то где V — объем раствора, мл
3. В единицах нормальности, т. е. числом грамм-эквивалентов (Э) вещества, растворенного в 1л раствора: где а — количество растворенного вещества, г; n — число грамм-эквивалентов; Если навеска исходного вещества (а) растворена в V миллилитрах, то где V — объем раствора, мл
Так как нормальность (N) раствора означает число грамм-эквивалентов вещества, содержащихся в 1л (или число миллиграмм-эквивалентов в 1мл) данного раствора, то, следовательно, произведение объема раствора, выраженного в литрах, на его нормальность равно числу грамм-эквивалентов вещества. Следовательно, если 1л данного раствора содержит N грамм-эквивалентов, то 1мл его содержит N/1000 грамм-эквивалентов или N миллиграмм-эквивалентов.
 4. Точным числом граммов растворенного вещества в 1мл раствора, т. е. в виде титра (Т): где а — навеска растворенного вещества, г; V — объем раствора, мл; Э — эквивалент растворенного вещества.
4. Точным числом граммов растворенного вещества в 1мл раствора, т. е. в виде титра (Т): где а — навеска растворенного вещества, г; V — объем раствора, мл; Э — эквивалент растворенного вещества.
Исходя из формул, можно написать следующие основные формулы:
 1) Вычисление нормальности:
1) Вычисление нормальности:
 где N — концентрация раствора, выраженная в единицах нормальности; а — навеска вещества, г; V — объем раствора, мл; Э — эквивалент исходного вещества; Т — титр раствора, г/мл.
где N — концентрация раствора, выраженная в единицах нормальности; а — навеска вещества, г; V — объем раствора, мл; Э — эквивалент исходного вещества; Т — титр раствора, г/мл.
2) Переход от нормальности к титру:
Билет №7
!!!!!! МЕТОД ИОДОМЕТРИИ основан на окислительно-восстановительных реакциях, связанных с превращением I2 в ионы Iˉ и обратно: I2 + 2 ē ↔ 2 I¯
Свободный йод является окислителем, а иодид-ион является восстановителем. Поэтому йодометрические методы применяются как для определения окислителей, так и для определения восстановителей.
Основными рабочими растворами в йодометрии являются растворы йода I2 для прямого титрования восстановителей и раствор натрия тиосульфата Na2S2O3·5H2O для определения окислителей и для обратного титрования восстановителей.
Основной титриметрической реакцией в методе йодометрии является взаимодействие раствора иода с рабочим раствором тиосульфата натрия:
I2 + 2 Na2S2O3 = 2 NaI + Na2S4O6 (тетратионат натрия)
В качестве индикатора в иодометрии используется водный раствор крахмала, который образует с молекулярным йодом иодкрахмальное соединение синего цвета. При титровании восстановителей рабочим раствором йода ТЭ определяется по появлению интенсивно-синего окрашивания. При титровании I2 рабочим раствором тиосульфата натрия конец реакции определяется по исчезновению синей окраски от одной капли раствора тиосульфата натрия. Крахмал необходимо добавлять в самом конце титрования, когда йода в растворе становится мало и раствор приобретает соломенно-желтый цвет.
Стандартизацию раствора Na2S2O3 с применением дихромата калия проводят методом замещения. К определенному количеству K2Cr2O7 прибавляют избыток иодида калия и кислоты, затем выделившийся I2 оттитровывают раствором Na2S2O3:
K2Cr2O7 + 6КI + 7H2SO4 = Cr2 (SO4)3 + 4K2SO4 + 7H2O + +3I2 2Na2S2O3 + I2 = Na2S4O6 + 2NaI
!!!!!! Рассмотрим обратимую химическую реакцию общего вида, в которой все вещества находятся в одном агрегатном состоянии, например, жидком:
аA + вB = сC + d D,
где A и B – исходные вещества прямой реакции; C и D – продукты прямой реакции; а, в, с, и d – стехиометрические коэффициенты.
В начальный момент времени, когда концентрация веществ A и B наибольшая, скорость прямой реакции также будет наибольшей и по закону действующих масс равна uпр = k1CАаCВв, где k1 – конс-та скорости прямой реакции.
С течением времени концентрация веществ A и B уменьшается, а, следовательно, уменьшается и скорость прямой реакции.
В начальный момент времени концентрация веществ C и D равна нулю, а, следовательно, и скорость обратной реакции равна нулю, с течением времени концентрация веществ C и D возрастает, а, следовательно, возрастает и скорость обратной реакции и она будет равна uобр = k2CCсCD d, где k2 – константа скорости обратной реакции.
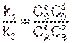 В момент достижения равновесия, концентрации принимают значение равновесных, а скорости равны между собой uпр = uобр, следовательно k1CАаCВв = k2CCсCD d
В момент достижения равновесия, концентрации принимают значение равновесных, а скорости равны между собой uпр = uобр, следовательно k1CАаCВв = k2CCсCD d
Перенесем константы скорости в одну сторону, а концентрации в другую:
 Отношение двух постоянных величин есть величина постоянная, и называется она константой химического равновесия:
Отношение двух постоянных величин есть величина постоянная, и называется она константой химического равновесия:
Константа равновесия показывает во сколько раз скорость прямой реакции больше или меньше скорости обратной реакции.
Константа равновесия – это отношение произведения равновесных концентраций продуктов реакции, взятых в степени их стехиометрических коэффициентов к произведению равновесных концентраций исходных веществ, взятых в степени их стехиометрических коэффициентов.
Величина константы равновесия зависит от природы реагирующих веществ и температуры, и не зависит от концентрации в момент равновесия, поскольку их отношение – всегда величина постоянная, численно равная константе равновесия. Если гомогенная реакция идет между веществами в растворе, то константа равновесия обозначается KС, а если между газами, то KР.

 где РС, РD, РА и РВ – равновесные давления участников реакции.
где РС, РD, РА и РВ – равновесные давления участников реакции.
Используя уравнение Клапейрона-Менделеева, можно определить связь между KР и KС рV = nRT
 Перенесем объем в правую сторону р = CRT Подставим уравнение для каждого реагента и упростим
Перенесем объем в правую сторону р = CRT Подставим уравнение для каждого реагента и упростим

где Dn – изменение числа молей газообразных участников реакции
Dn = (с + d) – (а + в) Следовательно,
KР = КС(RT)Dn
Из уравнения видно, что KР = КС, если не меняется количество молей газообразных участников реакции (Dn = 0) или газы в системе отсутствуют.
Необходимо отметить, что в случае гетерогенного процесса концентрацию твердой или жидкой фазы в системе не учитывают.
Константа равновесия имеет большое теоретическое и практическое значение. Численное значение константы равновесия позволяет судить о практической возможности и глубине протекания химической реакции.
Если K > 1, то данная реакция протекает со значительным выходом продуктов реакции; если K > 104, то реакция необратима; если K < 1, то такая реакция нетехнологична; если K < 10-4, то такая реакция невозможна.
Зная константу равновесия, можно определить состав реакционной смеси в момент равновесия и рассчитать константу выхода продуктов реакции. Константу равновесия можно определить, используя экспериментальные методы, анализируя количественный состав реакционной смеси в момент равновесия, или применяя теоретические расчеты. Для многих реакций при стандартных условиях константа равновесия – это табличная величина.
Влияние различных факторов на смещение химического равновесия отражает принцип Ле-Шателье (1884). Принцип Ле-Шателье применим как к гомогенным, так и к гетерогенным системам и дает качественную характеристику сдвига равновесия.
Аналитические определения можно проводить в химических системах, находящихся как в равновесном, так и в неравновесном состояниях. В первом случае концентрации веществ не меняются, поэтому продолжительность анализа несущественна и не влияет на выбор методики. Равновесные концентрации веществ связаны друг с другом через константу равновесия. Для химической реакции a A + b B  c C + d D эта константа равна
c C + d D эта константа равна
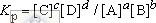
где в квадратных скобках указаны молярные концентрации соответствующих веществ, а показатели степени равны стехиометрическим коэффициентам уравнения химической реакции. Константы равновесия реакций, используемых в аналитической
химии, изменяются от 1 до 10100 и более. Многие методы анализа основаны на определении состояния равновесия. При анализе неравновесных систем определяют изменение концентрации реагирующих веществ во времени, т.е. скорость реакции. Она задается выражением 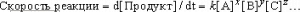
где k – константа скорости, [A], [B], [C] – молярные концентрации веществ A, B, C, сумма x, y, z – порядок реакции. Какие именно химические вещества фигурируют в уравнении скорости такой реакции и с какими степенями входят их концентрации, зависит от механизма химической реакции. При неравновесных определениях нужно успеть провести измерения за достаточно малое (по сравнению с продолжительностью самой реакции) время, так чтобы концентрации реагентов не изменились.
В аналитической химии есть несколько методов, основанных на определении положения химического равновесия. К ним относятся, в частности, классические гравиметрия и титриметрия, а также сравнительно новый иммунологический анализ.
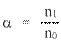 константа диссоциации Кд – это частный случай константы равновесия. Значения Кд для сильных электролитов часто бывают настолько велики, что их не удается измерить непосредственно. Помимо константы диссоциации, мерой силы электролита может служить степень диссоциации α. Это отношение числа распавшихся на ионы молекул (n1) к общему числу молекул (n0), изначально попавших в раствор:
константа диссоциации Кд – это частный случай константы равновесия. Значения Кд для сильных электролитов часто бывают настолько велики, что их не удается измерить непосредственно. Помимо константы диссоциации, мерой силы электролита может служить степень диссоциации α. Это отношение числа распавшихся на ионы молекул (n1) к общему числу молекул (n0), изначально попавших в раствор:
В зависимости от степени диссоциации различают электролиты сильные и слабые. Электролиты со степенью диссоциации больше 30% обычно называют сильными, со степенью диссоциации от 3 до 30% — средними, менее 3% — слабыми электролитами.
К сильным электролитам относятся почти все соли, некоторые кислоты и некоторые основания. К слабым электролитам относится большинство кислот (особенно органических) и оснований.
Степень диссоциации как сильных, так и слабых электролитов зависит от концентрации раствора (степень диссоциации тем выше, чем более разбавлен раствор). Более точной характеристикой диссоциации электролита является константа диссоциации, которая от концентрации раствора не зависит.
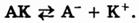 Выражение для константы диссоциации можно получить, если записать уравнение реакции диссоциации электролита АК в общем виде:
Выражение для константы диссоциации можно получить, если записать уравнение реакции диссоциации электролита АК в общем виде:
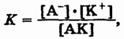 Поскольку диссоциация является обратимым равновесным процессом, то к реакции применим закон действующих масс, и можно определить константу равновесия как
Поскольку диссоциация является обратимым равновесным процессом, то к реакции применим закон действующих масс, и можно определить константу равновесия как
где К — константа диссоциации, которая зависит от температуры и природы электролита и растворителя, но не зависит от концентрации электролита.
Билет №8
!!!!!! В основе метода находятся реакции образования комплексных соединений (металло-лигандные реакции).
Метод комплексонометрии широко распространен для определения ионов Mg2+, Zn2+, Fe2+ и многих микроэлементов.
Комплексонометрия – титриметрический метод анализа, в основе которого лежит реакция взаимодействия определяемых ионов металлов с комплексонами. Комплексоны – аминополикарбоновые кислоты и их соли, способные образовывать сразу несколько связей с ионами металлов: ковалентные – Men+ с карбоксильными группами, донорно-акцепторные – Men+ с азотом аминогрупп.
Наибольшее значение из комплексонов имеет этилендиаминтетрауксусная кислота и ее двунатриевая соль (комплексон III или трилон Б – Na2H2T).
Взаимодействие катиона двухзарядного металла с трилоном Б можно представить схемой:

При титровании определяемых металлов, имеющих разные степени окисления, они связываются в бесцветные комплексонаты металлов, например:
Ме3+ + H2T2- = MeT- + 2H+
Ме4+ + H2T2- = MeT0 + 2H+
Полнота протекания реакции комплексообразования увеличивается по мере связывания ионов H+ в щелочной среде. Но, в некоторых случаях, требуется создание оптимального значения pH раствора, т.к. в избытке гидроксид-ионов могут образовываться нерастворимые гидроксиды определяемых металлов. Поэтому постоянство pH во время анализа поддерживается с помощью аммиачно-амонийного буферного раствора (NH4OH – NH4Cl).
Рабочим раствором в комплексонометрии может служить раствор трилона Б. Его чаще всего готовят приблизительной концентрации, а затем стандартизируют по растворам химически чистых хлорида или сульфата магния (MgSO4∙7H2O).
 ТЭ в комплексонометрии устанавливается с помощью металлохромных индикаторов.
ТЭ в комплексонометрии устанавливается с помощью металлохромных индикаторов.
Широко применяемым в комплексонометрии индикатором является эриохром черный Т, относящийся к группе азокрасителей и имеющий в молекуле хелатообразующие OH-группы
 Протон сульфогруппы в растворе диссоциирует практически полностью. Дальнейшее отщепление протонов от OH-групп приводит к изменению цвета индикатора. Окраска эриохром черного Т зависит от pH среды в растворе
Протон сульфогруппы в растворе диссоциирует практически полностью. Дальнейшее отщепление протонов от OH-групп приводит к изменению цвета индикатора. Окраска эриохром черного Т зависит от pH среды в растворе
Преобладающий в аммиачном буферном растворе анион Hind2- взаимодействует с ионами металла, образуя окрашенное в красный или фиолетовый цвет соединение:
M2+ + HInd2- (голубой) 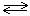 MInd- + H+.(красный)
MInd- + H+.(красный)
При титровании катиона M2+ ЭДТА в аммиачном буферном растворе в присутствии эриохром черного Т вблизи точки эквивалентности процесс протекает по уравнению
MInd- + H2Y2- + NH3 (красный)= MY2- + HInd2- + NH4+.(голубой)
В результате происходит изменение окраски раствора.
!!!!ОВР применяются в химическом анализе не только для открытия и разделения ионов, но лежат в основе методов оксидиметрии и использования редокс-индикаторов. ОВР широко используются в качественном и количественном анализе.
В качественном анализе ОВР используются для:
- переведения соединений из низших степеней окисления в высшие и наоборот;
- переведения малорастворимых соединений в раствор;
- обнаружени/удаления ионов;
Окислительно-восстановительные реакции широко используют не только в качественном анализе катионов, но и в анализе анионов.
Так в ходе анализа на анионы выполняют пробу на анионы-окислители (Cr2O72-, AsO43-, NO3-) действием раствора KI в кислой среде в присутствии хлороформа. При этом образуется свободный йод, окрашивающий слой хлороформа в красно-фиолетовый цвет.
Кроме того, проводят пробу на анионы-восстановители (C2O42-, S2O32-, S2-, SO32-, AsO33-, I-, NO2-), основанную на обесцвечивании раствора йода в слабокислой среде (за исключением AsO33-, которые обнаруживают в слабощелочной среде).
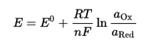 Уравнение Нернста — уравнение, связывающее окислительно-восстановительный потенциал системы с активностями веществ, входящих в электрохимическое уравнение, и стандартными электродными потенциалами окислительно-восстановительных пар. Уравнение Нернста позволяет предсказать максимальный рабочий потенциал, который может быть получен в результате электрохимического взаимодействия, когда известны давление и температура.
Уравнение Нернста — уравнение, связывающее окислительно-восстановительный потенциал системы с активностями веществ, входящих в электрохимическое уравнение, и стандартными электродными потенциалами окислительно-восстановительных пар. Уравнение Нернста позволяет предсказать максимальный рабочий потенциал, который может быть получен в результате электрохимического взаимодействия, когда известны давление и температура.
Рассмотрим окислительно-восстановительное равновесие
aARed + bBOx «aAOx + bBRed,
для которого можно записать полуреакции
aAOx + ne «aARed bBOx + ne «bBRed,
где n – число электронов, участвующих в ОВ–реакции.
Если компоненты этой системы находятся в химическом равновесии, то электродные потенциалы полуреакций равны (ЕА = ЕВ). Выразим электродные потенциалы полуреакций из уравнения Нернста, тогда при равновесии
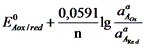  , ,
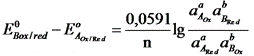 ,
,
, ,
,
,
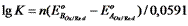 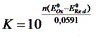 , ,
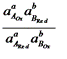  где n – число электронов, участвующих в ОВ – реакции.
Для аналитических целей используются реакции, которые протекают на 99,9 %, то есть: где n – число электронов, участвующих в ОВ – реакции.
Для аналитических целей используются реакции, которые протекают на 99,9 %, то есть:  В случае одноэлектронного перехода, когда n1 = n2 = 1, необходимая минимальная разность потенциалов для протекания реакции на 99,9 % следующая:
В случае одноэлектронного перехода, когда n1 = n2 = 1, необходимая минимальная разность потенциалов для протекания реакции на 99,9 % следующая:
 = 0,354В = 0,354В
|
Билет №9
!!!! Методы ОВТ, или редокс-методы,основаны на использовании реакций с переносом электронов — ОВР. Другими словами, ОВТ/редоксметрия, — это титрование, сопровождаемое переходом одного или большего числа электронов от иона-донора или молекулы (восстановителя)Red1к акцептору (окислителю) Ох2:
Red1+ Ох2=Ox1+Red2
Восстановленная форма одного вещества Red1, отдавая электроны, переходит в окисленную форму Ох1того же вещества. Обе эти формы oбpaзуют oдну peдoкc-пapу Oxl½Redl. Точто также и со второй парой.
В любой ОВР участвуют, по крайней мере, две редокс-пары.
Известно несколько десятков различных методов ОВТ. Обычно их классифицируют следующим образом.
Классификация по характеру титранта:
Оксидиметрия —методы определения восстановителей с применением титранта-окислителя;
Редуктометрия —методы определения окислителей с применением титранта-восстановителя.
Требования к реакциям ОВТ
1. Идут почти до конца
2. Быстрота реакции
3. Р-я протекает стехиометрически, без побочных р-ий
4. КТТ определяется точно и однозначно с И или без
В ОВ титровании, как и в КОТ, применяют прямое, обратное и заместительное титрование
|
|
|

История создания датчика движения: Первый прибор для обнаружения движения был изобретен немецким физиком Генрихом Герцем...

Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...

Архитектура электронного правительства: Единая архитектура – это методологический подход при создании системы управления государства, который строится...
Организация стока поверхностных вод: Наибольшее количество влаги на земном шаре испаряется с поверхности морей и океанов (88‰)...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!