

Кормораздатчик мобильный электрифицированный: схема и процесс работы устройства...

Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций...
Кормораздатчик мобильный электрифицированный: схема и процесс работы устройства...
Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций...
Топ:
Оценка эффективности инструментов коммуникационной политики: Внешние коммуникации - обмен информацией между организацией и её внешней средой...
Отражение на счетах бухгалтерского учета процесса приобретения: Процесс заготовления представляет систему экономических событий, включающих приобретение организацией у поставщиков сырья...
Интересное:
Отражение на счетах бухгалтерского учета процесса приобретения: Процесс заготовления представляет систему экономических событий, включающих приобретение организацией у поставщиков сырья...
Финансовый рынок и его значение в управлении денежными потоками на современном этапе: любому предприятию для расширения производства и увеличения прибыли нужны...
Уполаживание и террасирование склонов: Если глубина оврага более 5 м необходимо устройство берм. Варианты использования оврагов для градостроительных целей...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Ближайшие наши братья но крови - обезьяны. У одного из видов макаки, Macacus Rhesus, в эритроцитах был обнаружен белок, аналогичный такому же у человека. Белок назвали в честь макаки «резус-фактором», или Rh-фактором, по первым двум буквам ее видового названия. Открытие этого белка-антигена принадлежит двум американским ученым - А. Винеру и уже известному нам К. Ландштейнеру - и относится к 1940 году. (К этому времени К. Ландштейнер уже стал американским ученым.) Оказалось, что Rh-фактор есть в крови примерно 85 процентов людей земного шара, которые благодаря этому получили название «резус-положительных» (Rh+). Но у 15 процентов людей этого антигена нет, их назвали «резус-отрицательными» (Rh-). Таким образом, оказалось, что все люди разделены еще на две группы крови, и это объяснило неудачи переливания совместимых трупп, когда все необходимые правила соблюдались, но все же иногда наступали тяжелые осложнения и даже смерть.
Выяснилось, что в плазме крови резус-отрицательных людей нет антител против резус-фактора, и им можно спокойно переливать Rh- кровь. Но если им перелить даже небольшие количества Rh+ крови, то у них по отношению к антигену Rh начнут вырабатываться антитела. Если переливание такой крови будет однократным, ничего страшного не произойдет. Но повторное переливание Rh+ крови человеку, у которого уже появились антирезус-антитела, приведет к тяжелым осложнениям: эритроциты донорской крови быстро разрушатся.
Rh-фактор наследуется строго по законам Менделя, вернее, наследуются гены, определяющие синтез (или отсутствие синтеза) белка Rh. Ген отсутствия этого признака обозначается «rh». Резус-фактор является доминантным признаком. Поэтому резус-положительные люди могут быть либо гомозиготными (RhRh), либо гетерозиготными (Rhrh). Резус-отрицательный человек имеет рецессивный гомозиготный генотип1 rhrh. Иногда у резус-положительных родителей может родиться резус-отрицательный ребенок. Это произойдет тогда, когда родители гетерозиготны и от каждого из них ребенок получит по рецессивному гену rh. Вероятность рождения rh-отрицательного ребенка в этом случае составляет 25 процентов, то есть признак наследуется в отношении 3:1, по закону Менделя. У резус-отрицательных родителей никогда не может родиться резус-положительный ребенок.
|
|
Есть страны, где почти все население резус-положительное (например, в Африке, Японии, у индейцев Америки). У испанских басков, как правило, резус-отрицательная кровь. Предполагают, что резус-отрицательная кровь распространена среди народов Кавказа.
Для будущих детей резус-отрицательных женщин принадлежность к этой группе крови таит большую опасность. Если Rb- женщина забеременеет от Rh+ мужчины, то будущий ребенок может быть Rh+ или Rh-. Если он будет резус-отрицательный, то все будет хорошо. Но если он будет резус-положительным, то произойдет следующее. Как известно, плод и мать обмениваются компонентами крови через плаценту. Антигены Rh+ от плода станут поступать в Rh- кровь матери, в результате чего в крови матери появятся антирезус-антитела, которые неизбежно возвратятся в кровь плода, разрушат его эритроциты, костный мозг и другие ткани. Но так как эти антитела образуются в относительно поздние сроки беременности, то при первой беременности их будет мало, и они не успеют серьезно повредить ребенку. Поэтому обычно первый ребенок у Rh- женщины рождается здоровым.
Совсем другой финал может наступить при второй и последующих беременностях, если опять плод будет Rh+: анти-
1 Генотип-сумма всех генов организма, наследственная конституция.
резус-антитела станут быстро возникать в крови матери, количество их будет увеличиваться, так как часть их уже есть от предыдущих беременностей. Плод либо погибнет до рождения, либо родится больным гемолитической болезнью новорожденных (эритробластозом). Это заболевание характеризуется малокровием (анемией), общим отеком, поражением центральной нервной системы, желтизной кожных покровов. Если не применить экстренные меры, такие новорожденные погибают в первые 2-3 дня после рождения.
|
|
Каким образом удается спасти желтушного ребенка? В 1946 году американские ученые Валлерштейн, Винер и Вэкс-лер впервые спасли новорожденного, обреченного на смерть.
Им удалось заменить ему кровь. В самые ближайшие часы после рождения (не позже суток) они перелили ему 500-750 мл резус-отрицательной донорской крови, то есть 200-300 процентов объема по отношению ко всему объему крови ребенка.
Новорожденный был спасен. Этим методом спасли уже многих детей. После такого переливания состояние ребенка сразу улучшается, и он может полностью выздороветь. Опасность, которая грозит резус-отрицательной матери, заключается еще и в том, что если хотя бы раз она родила резус-положительного ребенка, она уже имеет в своей крови антирезус-антитела. Поэтому (если потребуется) ей можно переливать донорскую кровь только Rh-группы. Если женщине Rh-группы хотя бы однажды переливали кровь Rh+ группы, при повторном переливании такой же крови у нее могут развиться тяжелые осложнения, нередко оканчивающиеся смертью. Чем больше будет перелито Rh+ крови человеку резус-отрицательному (мужчине или женщине-безразлично), тем тяжелее могут быть последствия.
От 10 до 13 процентов всех браков заключается между Rh+ и Rh- людьми. Частота гемолитической болезни новорожденных по отношению ко всем бракам - 0,3-0,7 процента, сравнительно низкий процент, который объясняют тем, что не все женщины с Rh- кровью одинаково чувствительны к Rh+ антигенам плода. Заметная ответная реакция возникает у одной из 15-20 беременных. Почему это так, до сих пор не ясно. Ответную реакцию материнского организма
усиливают предыдущие беременности и аборты (если плод был Rh+) и, как уже говорилось, имевшие место переливания Rh+ крови.
Гемолитическая болезнь может возникать и в том случае, если плод и мать совместимы по резус-фактору. Это бывает тогда, когда у матери О группа. Как вы помните, группа крови О с другими группами несовместима. Описаны Rh+ семьи, в нескольких поколениях которых рождались дети с гемолитической желтухой. В разных случаях дети страдали по-разному, иногда переносили желтуху легко, в других случаях очень тяжело, с поражением центральной нервной системы.
|
|
О том, как часто встречается это заболевание, есть весьма разноречивые данные. Одни авторы пишут, что у одного новорожденного на 1000, другие - у трех на 7 000, третьи -у одного на 437 или даже на 150. Во всяком случае сейчас каждая мать может быть предупреждена, так как заранее определяются группы крови.
Рассказ о наследственных факторах крови мы закончим таблицей, наглядно показывающей, какую группу крови может унаследовать ребенок от своих родителей.
Комбинации генов, которые определяют у ребенка группу крови, отличающуюся от материнской, в таблице подчеркнуты.
Возможные варианты наследования группы крови плодом при разнообразных сочетаниях групп крови родителей
| Группа крови отца | ||||||||||||
| Группа крови матери | г, оо | II, А А | II, АО | III, ВВ | ш, во | JV, АВ | ||||||
| 1,00 | I, 00 | II, АО | I. 00 | Ш, ВО | ill, ВО | II, АО | ||||||
| И, АО | I, 00 | III, ВО | ||||||||||
| It, АЛ | II, АО | Ц, АА | II, АА | III, АВ | И, АО | II, АА | ||||||
| II, АО | IV, АВ | IV, АВ | ||||||||||
| П, АО | I, 00 | II, АО | II. 00 | Ш, ВО | I. 00 | U, АА | ||||||
| II, АО | II, АА | U, АО | IV, АВ | И, АО, | II, АО, | |||||||
| U, АА | IV, АВ | IV, АВ | ||||||||||
| ш, во | III, ВО | |||||||||||
| — | — | |||||||||||
| Ш;-ВВ | III, ВО | IV, АВ | III, ВО | III, ВВ | ш, во | Ш, ВВ | ||||||
| IV, АВ | Ш, ВВ | IV, АВ | ||||||||||
| Группа ярони отца | ||||||||||||
| Грушга кров» матери | Г, 00 | II, АЛ | П, АО 1П, ВВ | пг, во | IV, АВ | |||||||
| ш, во | Г, 00 | II, АО, | Г, 00 | III, ВО | I, 00 | II, АО^ | ||||||
| - | Ш, ВВ | |||||||||||
| ш, во | IV, АВ | Н, АО | Ш, ВВ | III, ВО | ш, во | |||||||
| ш, во | Ш, ВВ | IV. АВ | ||||||||||
| IV, АВ | ||||||||||||
| IV, АВ | 11, АО | П, АА | II, АО | III, ВВ | II, АО | Н, АА | ||||||
| ш, во | IV, "АВ | III, В© | IV, АВ | 111, ВО | IV, АВ | |||||||
| III, AA | IV, АВ | Ш, ВВ | ||||||||||
| IV, АВ | III, ВВ | |||||||||||
ГЕНЕТИКА ЧЕЛОВЕКА
|
|
Мальчик или девочка? Как хочется заранее получить ответ на этот вопрос, в семье, где уже есть дочь и жаждут сына или, наоборот, где два-три братика ждут сестричку. Но главные волнения не об этом: кто бы ни родился, сын или дочь,-был бы здоровым.
Плацентарный барьер становится на пути к радостной и здоровой жизни первым барьером, который надо преодолеть. О неприятностях, связанных с несовместимостью крови, мы рассказали. Могут быть и другие препятствия к нормальной и здоровой жизни ребенка. По ряду стран существует весьма печальная статистика. Например, в США из 5-10 миллионов зачатий половина завершается гибелью эмбрионов на ранних стадиях развития. Из 3,2 миллиона эмбрионов, достигших 20-недельного возраста, около 40 000 погибает, не успев родиться. Почти такое же количество новорожденных умирает в первый месяц после рождения от того, что они родились с какими-нибудь отклонениями в развитии тканей, органов или систем. Примерно 40 000 новорожденных остаются жить с врожденными пороками, которые иногда можно лечить. Каждый год рождается не менее 90 000 умственно отсталых детей и 150 000 детей, которые будут с трудом учиться. Они дополнят и без того тревожную картину судьбы будущих американцев. В чем же причина этих несчастий? Отчасти -в дефектных генах, передающихся по наследству.
В настоящее время около 6 процентов всех людей имеют какие-либо отклонения от биологической нормы. Каждый здоровый человек в хромосомном аппарате своих клеток содержит не менее 12 дефектных генов, которые себя никак не проявляют. Вместе с тем известны сотни наследственных болезней, с которыми ребенок появляется на свет. Значит, они возникли еще в период внутриутробного развития. И тогда родители начинают ломать голову: откуда бы это могло быть? Анализируют биологические особенности своего организма, считая себя, свою наследственность виновной в этом. Порой ищут причину в поведении женщины во время беременности, вспоминают, не было ли у нее испуга, физических перегрузок, инфекционных или иных болезней.
На вопрос: где причина врожденных болезней? - пытался ответить еще Каспар Фридрих Вольф. Он писал в «Теории способности передачи потомству», что «... в семьях шестипалых шестипалый отец производит на свет сына также шестипалого или от гермафродита родится гермафродит». И если принять во внимание, что «...у человека формируется пять пальцев, две руки и две ноги исключительно потому, что у родителей было столько же пальцев, столько же рук и столько же ног...», то разве нельзя убедиться, что «именно структура родителей является причиной структуры потомства»? На первый взгляд это действительно так. Но Вольф уже тогда понимал сложность механизма передачи признаков потомству, ибо вслед за этими факторами он анализирует и другие, критикуя сложившееся неверное представление о том, что «сами части тела являются причиной формирования частей эмбриона или имеют влияние на эти формирующиеся части». Он приводит такие доводы: «Люди, у которых ампутированы ноги и кисти рук или полностью руки, люди с укороченным носом или какой-либо другой частью тела, слепые, горбатые, люди, у которых какие-нибудь другие части тела поражены болезнями, тем не менее рождают здоровых и неповрежденных детей».
|
|
В XVIII веке русский академик еще не мог разграничить наследственные и ненаследственные болезни. О генах тогда и речи не было. Однако о наследовании некоторых болезней было известно давно. Так, в 1716 году у здоровых
родителей родился Эдвард Ламберт, кожа которого быстро потемнела, а затем покрылась чешуйками. У него было 6 сыновей тоже с кожей дикобраза, причем признак повторялся в шести последующих поколениях у мальчиков. История знает передачу по наследству врожденной ночной слепоты, которую наследовали на протяжении многих поколений 134 потомка одного знатного рода.
Существовало предание, что сифалангию (сращивание суставов пальцев), прослеженную на протяжении длительного времени в некоторых родах, происходящих от знаменитого полководца Столетней войны Джона Тальбота, потомки унаследовали именно от него. При ремонте гробницы Тальбота в 1917 году удалось доказать, во-первых, что находящийся в ней скелет действительно принадлежал знаменитому полководцу, так как на нем обнаружили следы предсмертных ранений, полученных им под Шатильоном и описанных в хрониках того времени. Во-вторых, на скелете была найдена сифалан-гия, которая передавалась по наследству на протяжении 14 поколений. Описано множество болезней, стойко передающихся по наследству. Среди них - глухонемота, подагра, шизофрения, некоторые формы диабета, мраморная болезнь (ломкость костей), альбинизм (бесцветные кожа, волосы, розовые глаза, радужные оболочки которых лишены красящего пигмента, благодаря чему просвечивающие кровеносные сосуды делают их розовыми), раннее поседение и полысение и многие другие.
Если болезнь новорожденного была вызвана воздействием на организм плода каких-нибудь неблагоприятных факторов внешней среды, она считается приобретенной. Если она обусловлена дефектными генами родителей - наследственной.
Для новорожденного безразлична причина возникновения его болезни. Но родителям ее знать необходимо. Ведь если патология наследственна, она может повториться у будущих детей. Всегда ли? И на этот вопрос во многих случаях уже найден ответ.
Согласно первому закону Менделя, у первого поколения гибридов вероятность появления гетерозиготы (организма, объединившего гены разных признаков двух родителей, способного потом дать несколько типов генетически различных
половых клеток) равна 100 процентам. Вероятность того, что гибриды будут обладать доминантным признаком, также равна 100 процентам. (Вспомните, как преобладали гены А и В в сочетаниях АО и ВО групп крови ребенка. На этом примере было хорошо видно, что вероятность появления скрытого, рецессивного признака (гена О) равнялась нулю).
Иначе выглядит прогноз в отношении потомства второго гибридного поколения: у внуков в 25 процентах случаев проявится один доминантный ген, в 25 процентах - рецессивный, а в 50 процентах хромосомы внуков будут содержать оба гена, то есть будут гетерозиготны. (Помните, у Менделя 1:2:1.) У 50 процентов гетерозиготных внуков когда-нибудь родятся правнуки. У них интересующие нас признаки будут наследоваться в отношении 3:1 (три четверти потомков унаследуют доминантный ген, и одна четверть - рецессивный). Вроде бы все легко и просто, но не все так хорошо укладывается в схему, когда дело касается людей. Частота проявления действия генов у человека оказалась менее 100 процентов. Если же по законам Менделя ожидается, что ген проявится у половины потомков, то лишь половина от этой половины потомков, имеющих этот ген, будет обладать признаком, им определяемым.
Человек многим отличается от других живых существ, прежде всего - развитием мозга и зависимостью от социальной среды. Поэтому возникла ветвь генетики - медицинская генетика, или генетика человека, учитывающая все достижения современной науки и все особенности объекта Homo sapiens.
С людьми нельзя поступать так, как с разными сортами гороха,- метод гибридизации здесь неприемлем. У исследователя наследственных болезней есть другие способы. Так, из многих семей он может отобрать такие, в которых прослеживается наследование (не обязательно заболевания) любого признака, отличающего эту семью от других.
Например, несколько членов Габсбургской династии королей имели выступающую нижнюю челюсть и особым образом измененную нижнюю губу.
Наследование этих признаков было изучено очень тщательно, и результаты, вместе с историческими портретами, опубликованы Императорской академией, находившейся под покровительством самой семьи Габсбургов. Присмотревшись к
портрету члена семьи XIV века и портрету потомка, жившего в XIX веке, можно видеть, что этот признак передавался из поколения в поколение сквозь столетия и воспроизводился в точности.
(Маленькая историческая справка: германские короли династии Габсбургов правили до 1806 г. так называемой Священной Римской империей германской нации, Испанией - с 1516 до 1700 г., Австрийской империей - с 1804 г., Австро-Венгрией-с 1867 по октябрь 1918 г. Первый император этой династии - Рудольф I - царствовал с 12.73 по 1291 г. Последний - Карл I - правил всего два года (1916-1918) и был свергнут национально-освободительным и рабочим движением в Германии 1918 г.)
...Сложность генетических исследований среди людей заключается в том, что далеко не все хранят портреты своих предков с десятого (или дальше) колена и даже не знают ни фамилий, ни имени их. Исследования требуют большого времени и создают необходимость изучать многих людей.
Так как в основном исследования родословной строятся на рассказах о родственных связях, они не всегда бывают точными. Да и в некоторых семьях дети могут не знать, что их воспитывал приемный отец или приемная мать.
Даже многодетные семьи не обладают достаточным материалом, чтобы можно было делать выводы о наследовании того или иного признака. И все же изучение наследственных болезней у трех поколений одной семьи (иногда удается анализировать и четвертое - правнуков) позволяет судить о том, является ли болезнь наследственной или возникла под влиянием факторов окружающей среды. С этой же целью выявляют одинаковые признаки у близнецов. У однояйцевых близнецов, развившихся из одной зиготы,- одинаковый набор генов. Поэтому их общие признаки почти всегда имеют наследственную природу. Если у таких близнецов существуют различия, их объясняют влиянием факторов окружающей среды.
Близнецы ~ самый настоящий клад для генетики. Недаром в годы второй мировой войны близнецов берегли как государственное сокровище в оккупированной немцами Дании и в Швеции. Реестр датских и шведских близнецов охранялся, как золотой фонд. Сравнивая, как и чем они болеют, как реагиру-
ют на лекарства, как ведут себя при разных обстоятельствах, можно выяснить, что наследственно, а что может изменяться под влиянием среды. Так как у разнояйцевых близнецов гораздо меньше общих генов, интересно для выявления наследственных болезней сравнивать их с однояйцевыми.
Как правило, медицинский генетик может проследить характер наследования признаков лишь в одном или двух поколениях, так как его собственная жизнь ограничена определенным периодом, так же как и его деятельность в области генетики. Поэтому медицинская генетика развивается не так быстро, как хотелось бы.
Однако в настоящее время у людей уже выявлено много доминантных признаков. Если зигота, из которой разовьется мальчик или девочка, содержит два разных гена, отвечающих за цвет глаз - голубой и карий, у ребенка будут карие глаза, так как ген кареглазости доминирует.
Точно так же преобладающими признаками являются: короткие пальцы на руках (брахидактилия), неспособность различать предметы при плохом освещении («куриная слепота»), значительно укороченные конечности при нормальном туловище и голове, выступающая вперед нижняя челюсть (в роде Габсбургов), повышенное содержание холестерина в крови, ямочки на щеках, свободная - неприросшая мочка уха, способность свертывать язык трубочкой, большие глаза, прямой разрез глаз, близорукость, крупный нос, широкие ноздри, длинный подбородок, волосы с мелкими завитками, поседение волос к 25 годам, раннее облысение у мужчин, наличие зубов при рождении, смуглая кожа, веснушки, нормальный рост, праворукость, сопрано у женщин и бас у мужчин, абсолютный музыкальный слух, склонность к ожирению.
Рецессивными признаками будут: отсутствие волос, тонкий большой палец, приросшая мочка уха, голубые глаза, сахарный диабет юношей, маленькие глаза, нормальная острота зрения, широкий и прямой нос, короткий подбородок, вьющиеся, волнистые или прямые волосы, поседение волос после 40 лет, облысение у женщин, отсутствие зубов при рождении, белый цвет кожи, длинный череп, отсутствие склонности к ожирению, леворукость, альт у женщин и тенор у мужчин, наследственная глухота.
Около 93 процентов болезней, подкарауливающих младенца с момента появления на свет, объясняются наследственным предрасположением - своеобразием в генном наборе, включающим передающийся по наследству дефект. До поры до времени он может не проявляться вообще, как бы притаившись в ожидании подходящих условий. Чаще всего это различные нарушения обмена веществ или биосинтеза белка. В настоящее время лучше других изучены причины нарушений углеводного обмена. Если из-за нарушений в генном аппарате в клетке отсутствует один из ферментов, необходимых для углеводного обмена,- гликозидаза, то углеводы не расщепляются и начинают накапливаться. Возникает так называемая болезнь накопления, из-за которой новорожденный погибает в возрасте 1-3 месяцев.
Это как раз тот случай, когда и отец и мать здоровы, но в их хромосомах есть дефектный рецессивный ген, доставшийся им в наследство от предков. Дефект не проявлялся ни у отца, ни у матери потому, что в паре с дефектным геном доминировал ген нормального синтеза гликозидазы. Если ребенок гомозиготен по рецессивному гену, он будет нежизнеспособным. Единственный пока способ помочь родителям - установить диагноз до рождения и своевременно прервать беременность.
Нарушения в углеводном обмене серьезны потому, что от углеводов зависит контакт между клетками; клетки «узнают» одна другую благодаря углеводам, расположенным близко к их поверхности. Углеводы определяют специфику веществ, от которых зависит группа крови, они поставляют клетке необходимую энергию и участвуют во многих других жизненно важных процессах.
В суставах, костях, хрящах, связках, соединительнотканных волокнах сердца и сосудов есть углеводы, называемые сейчас, гликозаминогликанами (еще недавно их называли муко-полисахаридами). Если нарушается их обмен, дети отстают в умственном и физическом развитии. У них резко замедлен рост, деформированы грудная клетка и конечности, часто бывает горб. Как правило, эта болезнь прогрессирует, лечить ее очень трудно и в большинстве случаев - безуспешно.
Предполагают, что одним из заболеваний этой группы страдал Никколо Паганини. Как сообщил недавно один из аме-
риканских научных медицинских журналов, несколько странный внешний облик великого скрипача получил объяснение у современных генетиков. Мертвенно-бледное, как бы вылепленное из воска лицо, глубоко запавшие глаза, сверхгибкие и невероятно длинные пальцы, угловатость движений - все это характерно для синдрома Марфана, наследственной болезни, впервые описанной через 56 лет после смерти Паганини. Необычным строением пальцев объяснял виртуозность игры Паганини Гете. Но конечно же, не только строение пальцев - плюс к этому Паганини, несомненно, обладал способностями музыкального гения.
И другие нарушения обмена белков, жиров, аминокислот, минеральных веществ тоже могут быть наследственными. Генетически обусловленных нарушений обмена сейчас известно около 2000. Лишь немногие из них можно лечить.
Несколько лет назад в нашей печати было опубликовано сообщение из Института экспериментальной медицины АМН СССР, в котором говорилось о попытках исправлять последствия генных нарушений в организме животных. Дело в том, что наследственная информация, полученная от родителей, реализуется не сразу. В ходе развития зародыша можно выделить несколько критических моментов. Из них главнейшими являются два: когда устанавливается связь между эмбрионом и организмом матери и когда начинается морфогенез.
Нарушения жизнедеятельности эмбриона в первый критический период оканчиваются, как правило, его гибелью. Это, несомненно, меньшее зло, чем рождение урода, если нарушения возникнут позже.
Развивающийся человеческий организм особенно чувствителен к повреждениям на 1,3 и 9-й неделях внутриутробной жизни. Возможно, если в это время воздействовать на зародыш извне, можно будет лечить врожденные дефекты.
Опыты проводились вначале на плодовых мушках-дрозофилах, потом на мышах. Хотя глубокие причины наследственных «поломок» находились в хромосомном аппарате, оказалось, что реализация их зависит от условий, в которых шло развитие зародыша.
Прогревание при 32-34° приводило к тому, что у наследственно короткокрылых мух потомство имело почти нормальные крылья.
Чистая линия мышей с недоразвитыми глазами производила мышат с глазами большего размера, если в рацион беременных самок добавляли некоторые витамины или короткое время прогревали их при температуре около 40°.
Уменьшали наследственные нарушения антибиотики и рациональный пищевой режим. Таким образом, дело не так безнадежно, как кажется. Но от экспериментов на животных к лечению человека- долгий и нелегкий путь.
Некоторые наследственные болезни уже лечат. Например, гемофилию. В крови людей, больных гемофилией, перестает вырабатываться либо фибриноген - высокомолекулярный белок, свертывающийся при образовании раны, либо тромбин -фермент, способствующий свертыванию фибриногена. Значит, стоит ввести в организм недостающие вещества, и больной уже не рискует погибнуть от кровотечения.
Действенный метод борьбы с наследственными болезнями - генетическая консультация. Будущим родителям, после того как устанавливаются наследственные нарушения у них или у их предков, разъясняется степень риска рождения у них больного ребенка. Риск во много раз выше, если заключается близкородственный союз - между двоюродными братьями и сестрами, так как в их генетическом аппарате могут быть одинаковые дефектные гены. У детей, родившихся от таких браков, гораздо чаще встречаются наследственные болезни и врожденные уродства. Кровное родство родителей на пять процентов повышает смертность новорожденных. Правда, эта закономерность проявляется лишь в двух ближайших поколениях. Если проанализировать родственные связи на пять и более поколений назад, то есть построить генеалогическое дерево, может быть получен самый неожиданный результат: в родстве окажутся те, кто об этом и не подозревал.
За последние 100 лет число родственных браков сократилось, так как исчезают расовые, национальные и социальные предрассудки, развиваются культурные связи между странами и народами. Чем больше расстояние между местами постоянного жительства семей каждого из родителей, тем меньше у родителей одинаковых генов, среди которых могут обнаружиться дефектные.
7. «РОДИЛА ЦАРИЦА В НОЧЬ...»
В 1886 году английский невропатолог Л. Даун впервые описал врожденную болезнь, поражающую, в среднем, одного из 600 новорожденных. Больные дети - вялые, с толстым, неповоротливым языком, расплющенным носом, узкими щелочками глаз. Часто у них врожденные пороки сердца и всегда слабоумие. Значительная часть из них - контингент психиатрических лечебниц.
По имени врача, описавшего болезнь, ее до сих пор называют синдромом Дауна. Долго болезнь Дауна считали результатом повреждения плода во время внутриутробного развития. Это оказалось неверным - истинную причину болезни открыл французский ученый Жером Лежен. Он посмотрел под микроскопом множество клеток, взятых у больных детей, и обнаружил, что в них не 46, а 47 хромосом.
Это может быть потому, что в организме женщины при образовании яйцеклетки во время мейоза расхождения какой-либо пары хромосом не произошло. И тогда в яйцеклетку попадут две одинаковые хромосомы. В данном случае -

Схема плацентарного барьера в конце 40-й недели внутриутробного развития. Кровеносные сосуды значительно приближены к материнской крови, так как плацентарный барьер истончен. Отсутствует слой В, а все сосуды лежат в зоне, максимально приближенной к поверхности ворсинок.
хромосомы 21-й пары. После оплодотворения их будет уже не две, а три, так как добавится 21-я хромосома отца. Поэтому нарушение еще называют трисомией 21 или нерасхождением по 21-й хромосоме. С возрастом матери повышается риск рождения «дауна»: заметили, что у женщин от 19 лет до 21 года на 2500 детей рождается один «даун», а у женщин 45 лет - один на 40 детей. Естественным было предположить, что сдвиги в механизме мейоза зависят от гормональных изменений, наступающих с возрастом в организме женщины. Но сейчас известны другие причины. Могут быть нарушены первые этапы дробления зиготы, и тогда одна 21-я хромосома присоединяется к другой (к 15-й или 22-й). Это явление называют транслокацией хромосом. В первом случае (то есть когда в яйцеклетке лишняя 21-я хромосома) болезнь Дауна не наследуется, хотя мать может иметь несколько «даунов». Во втором случае аномалия будет передаваться из поколения в поколение, дети «даунов» окажутся их точной копией, но не все, как это выяснил анализ

Схема плацентарного барьера при гибели плода до рождения. Нарушено развитие хориапьных ворсинок. Кровеносные сосуды плода расположены на значительно большем расстоянии, чем в норме, так как имеются не только в зоне I, но и во II и III.
родословной семьи с синдромом Дауна, обусловленным транслокацией хромосом1.
«Дауны» живут по нескольку десятков лет. Лечение их успеха не приносит. Иногда их удается научить читать и писать.
Если в 21-й паре хромосом одна больше другой (это бывает, когда одна из хромосом потеряла значительную часть своего плеча2), развивается злокачественная болезнь крови -миелоидная лейкемия.
Нерасхождение по 18-й хромосоме (синдром Эдвардса) всегда смертельно. Дети умирают через несколько месяцев после рождения. Они рождаются со множеством дефектов во внутренних органах. У них маленькие глаза, неправильно расположенные уши, короткая грудина, врожденные пороки сердца, недоразвитая скелетная мускулатура, отсутствие шеи, дефекты пальцев. С синдромом Эдвардса рождается один ребенок из 6500. Девочки с синдромом Эдвардса рождаются в два раза чаще, чем мальчики. Как в случае трисомии 21, трисомия 18 зависит от возраста матери: чем старше мать, тем больше вероятность нерасхождения 1-8-й пары хромосом.
Под названием «синдром Патау» известна трисомия 13 (нерасхождение по 13-й паре хромосом). Эта аномалия бывает у одного новорожденного из 4600. У детей при синдроме Патау не срастаются верхняя губа и верхнее небо. (При нормальном развитии эти части закладываются симметрично с двух сторон и срастаются посредине.) В народе такие аномалии развития издавна называют «волчьей пастью» и «заячьей губой». Они сопровождаются врожденными пороками сердца и увеличением числа пальцев до шести (полидактилией). Помимо перечисленных, встречаются и другие нарушения - в развитии ушей, половых желез и т. д. Дети с трисомией 13 рождаются с малой массой тела (менее 2500 г) и погибают, как правило, в первые месяцы жизни.
«Заячья губа» и «волчья пасть» бывают у новорожденных и тогда, когда хромосома 4-й пары частично утратила свое короткое плечо. Такая же аномалия в хромосоме 5-й пары
1 Ш. Ауэрбах, Проблемы мутагенеза. М„ Мир, с. 424, схема, с. 425. 1 Плечи хромосомы - расстояния от точки, к которой к хромосоме прикрепляется нить центриоли во время деления клетки до ее конца.
приводит к появлению синдрома «кошачьего крика» - этот специфический крик возникает при недоразвитии голосового аппарата ребенка.
Интересно, что если, кроме двух обычных хромосом 13-й пары, есть добавочное верхнее плечо от третьей 13-й хромосомы, то это не приводит к патологии.
В литературе сообщалось о канадской семье, в которой молодая женщина в 21 год имела двоих детей и ожидала третьего. У одного из детей была полидактилия, и мать обратилась к врачам с просьбой поставить диагноз до рождения, так как беспокоилась, что третий ребенок родится с таким же уродством. При обследовании оказалось, что развивается девочка, у которой плюс к паре хромосом 13 есть верхнее плечо такой же 13-й хромосомы. Добавочное плечо 13-й хромосомы было обнаружено в клетках матери и бабушки (по материнской линии). Так как и мать, и бабушка были здоровы, а генетикам было известно уже более ста случаев, когда у совершенно здоровых людей обнаруживали такое же нарушение в хромосомном аппарате, врачи порекомендовали не прерывать беременности.
Родилась здоровая девочка. Когда ей исполнилось 10 месяцев, ее клетки исследовали и нашли в них добавочное плечо 13-й хромосомы, не причинившее ей никакого вреда.
Такие наблюдения заставляют задуматься: очевидно, медицинская генетика еще далеко не все знает. На пути от генов до признаков скрыто много неожиданного и неизвестного...
Множественные уродства бывают у детей, в клетках которых обнаруживается аномальная 18-я хромосома, утратившая свое длинное или короткое плечо. Аномалии лю-

Схематическое изображение двух хромосом из разных пар после дифференциального окрашивания. Видны чередующиеся светлые и темные диски разной ширины, создающие специфику для каждого плеча хромосомы.
бой пары хромосом от 1-й по 22-ю, как правило, смертельны, и развитие плода самопроизвольно прерывается (спонтанный, или самопроизвольный, аборт).
Из общего количества беременностей 30-40 процентов заканчивается спонтанным абортом, и изучение хромосом в тканях абортусов (плодов, прекративших развитие) показало, что почти половина из них имеет аномалии в хромосомах. Чаще всего среди них обнаруживаются трисомии. Их опознают с помощью дерматоглифики (нерасхождения по 21-й, 18-й и 13-й хромосомам меняют рисунки кожи пальцев рук). Встречаются также добавочные наборы хромосом - трипло-идные или тетраплоидные клетки, отсутствие одной Х-хро-мосомы. Другие нарушения в хромосомном аппарате встречаются реже.
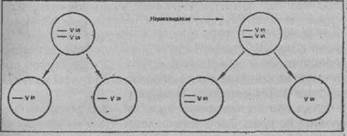
Схематическое изображение перераспределяющихся хромосом при мейозе. Слева: в норме в каждой зрелой половой клетке остается по одной хромосоме из каждой пары. Справа: иллюстрация к нерасхождению хромосом, когда в зрелой половой клетке появляются обе хромосомы из какой-либо пары
Изменения в числе половых хромосом не грозят жизни новорожденного, а проявляют себя иначе. Если в яйцеклетке произошло нерасхождение по Х-хромосоме, то после оплодотворения мужской гаметой с Y-хромосомой в ней будет не 46, а 47 хромосом, причем последняя пара будет представлена тройкой XXY. Такое сочетание половых хромосом определит у новорожденного синдром Клайнфельтера, редкое заболевание, встречающееся только у мужчин, да иначе и не может быть, так как Y-хромосома определяет мужской пол. (Правда, ее определяющая роль при избыточных Х-хромосомах не так сильна.)
У больных синдромом Клайнфельтера полова
|
|
|
Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций...

Таксономические единицы (категории) растений: Каждая система классификации состоит из определённых соподчиненных друг другу...
Типы сооружений для обработки осадков: Септиками называются сооружения, в которых одновременно происходят осветление сточной жидкости...

Опора деревянной одностоечной и способы укрепление угловых опор: Опоры ВЛ - конструкции, предназначенные для поддерживания проводов на необходимой высоте над землей, водой...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!