II начало термодинамики, как физическая закономерность отличается от первого начала термодинамики. Закон сохранения энергии выполняется одинаково, как для макроскопических тел, так и для отдельных атомов и молекул. Опыты показывают, что II начало термодинамики применимо к системам, которые содержат большое количество молекул (частиц).
При применении II начала термодинамики к объектам, которые содержат малое число молекул (частиц) наблюдаются отклонения от этого закона.
Например, броуновское движение не подчиняется II началу термодинамики, оно противоречит II началу термодинамики. Броуновская частица получает энергию от молекул окружающей среды (жидкости), температура которой равна температуре броуновской частички. При этом нарушается II начало термодинамики: теплота переходит в работу без каких-либо изменений в окружающей среде.
Это объясняется тем, что II начало термодинамики – статистический закон. Явления, которые происходят в макроскопической системе, складывающейся из большого числа микрочастиц, носят статистический характер.
Статистический подход ко II началу термодинамики основан на применении для оценки того или иного состояния системы понятия вероятности.
Состояние системы, которое характеризуется ее термодинамическими параметрами, называется макроскопическим состоянием.
Состояние системы, которое характеризуется состоянием каждой ее молекулы (частицы), называется микроскопическим состоянием.
Каждое микроскопическое состояние системы характеризуется определенной вероятностью. Молекулы газа двигаются хаотично, поэтому существует много возможных микроскопических состояний, соответствующих определенному макроскопическому состоянию системы. Если считать, что все микроскопические состояния равновероятны, то можно найти вероятность данного макроскопического состояния.
Однако такой способ оценки вероятности того или иного макроскопического состояния достаточно сложный.
Поэтому в статистической физике используют термодинамическую вероятность, или статистический вес макроскопического состояния.
Термодинамическая вероятность W макроскопического состояния – это число способов (число микроскопических состояний), которыми может быть осуществлено данное макроскопическое состояние.
В отличие от математической вероятности, которая не может быть больше единицы, термодинамическая вероятность всегда много больше, либо равна единице.
Рассмотрим газ, который состоит из четырех молекул с номерами 1,2,3,4. Пусть этот газ находится в половине A сосуда с перегородкой. Другая половина сосуда B – пустая (рис.1).

Рис.1. Распределение молекул между двумя половинами сосуда
Если удалить перегородку, то молекулы, двигаясь хаотично, могут оказаться в разные моменты времени как в половине A, так и в половине B данного сосуда. Поэтому в разные моменты времени будут возникать различные распределения молекул между половинами A и B сосуда: поровну, три слева одна справа, и т.д.
Подсчитаем число способов распределения молекул между двумя половинами сосуда (табл.1)
Таблица 1:
| Состояние
| Способ реализации
состояния
| Число способов реализации данного состояния (термодинамическая вероятность W)
|
| Число молекул, A
| Число молекул, B
| №,№ молекул
A
| №,№ молекул
B
|
|
|
| -
| 1,2,3,4
|
|
|
|
|
| 2,3,4
|
|
|
| 1,3,4
|
|
| 1,2,4
|
|
| 1,2,3
|
|
|
| 1,2
| 3,4
|
|
| 1,3
| 2,4
|
| 1,4
| 2,3
|
| 2,3
| 1,4
|
| 2,4
| 1,3
|
| 3,4
| 1,2
|
|
|
| 1,2,3
|
|
|
| 1,2,4
|
|
| 1,3,4
|
|
| 2,3,4
|
|
|
|
| 1,2,3,4
| -
|
|
| Всего способов:
| 24 = 16
|
Каждая молекула с равной вероятностью может оказаться, как в половине A, так и в половине B сосуда. Следовательно, каждое из 16 распределений молекул между половинками сосуда может осуществляться одинаково часто.
Поэтому число способов реализации данного состояния определяет вероятность данного состояния. Как видно из таблицы наибольшее число способов распределения осуществляет такое состояние, при котором и слева и справа будет по две молекулы, т.е. равномерное распределение молекул между двумя половинами сосуда: число способов размещения – 6, математическая вероятность 1/6.
Может случиться, что все 4 молекулы опять окажутся в половине A сосуда. Это значит, что газ, который расширился, самопроизвольно сжался. Расширение газа в пустоту оказывается обратимым процессом, что противоречит II началу термодинамики. Однако только одно распределение молекул из 16 создает такое состояние, при котором все молекулы соберутся в половине A сосуда.
Математическая вероятность такого события 1/16 – мала по сравнению с вероятностями других размещений, но не очень, при небольшом общем числе молекул.
Как видно из таблицы общее число способов распределения n молекул жду двумя половинами сосуда – 2 n. В реальных системах число молекул очень велико. В 1см3 воздуха при нормальных условиях, находится ~ 1020 молекул. Общее число способов распределения молекул между двумя половинами сосуда (V = 1см3) равно:
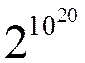
И, только один из этих способов будет соответствовать тому, что все молекулы соберутся в половине A этого сосуда.
Такое событие возможно, однако его математическая вероятность будет равна:

Эта вероятность настолько мала, что практически осуществиться не может. И наоборот, число способов, которыми осуществляется равномерное распределение молекул в объеме сосуда значительно больше по сравнению с любым другим распределением.
Состояние равномерного распределения молекул по объему сосуда наиболее вероятное и является равновесным.
Отсюда следует, что процесс самопроизвольного сжатия газа очень маловероятный. Иначе говоря, процесс расширения сжатого газа на весь сосуд необратимый, потому что обратный процесс самопроизвольного сжатия газа очень маловероятный.
Обобщая вышеизложенное можно сделать вывод: всякий необратимый процесс – это такой процесс, обратный которому очень маловероятный.
Статистический характер II начала термодинамики хорошо выявляется в формулировке Больцмана:
Природа стремится от менее вероятных состояний к более вероятным.
Из всех видов движения наиболее вероятным является хаотическое (тепловое) движение молекул. Поэтому любое упорядоченное движение молекул всегда стремится перейти в неупорядоченное.
При выполнении работы происходит переход макроскопического, упорядоченного движения тела, под действием внешних сил в неупорядоченное, хаотическое движение молекул (нагревание тел из-за сил трения). Этот переход более вероятен. Получение работы за счет теплоты означает переход хаотичного движения молекул в упорядоченное движение макроскопического тела. Такой переход менее вероятен.
II начало термодинамики показывает необратимость перехода работы в теплоту. Это значит, что обратный переход теплоты в работу соответствует переходу от более вероятного состояния к менее вероятному.
Если изолированная система находится в состоянии с максимальной энтропией, которая соответствует состоянию с максимальной вероятностью, то в ней не могут самопроизвольно протекать какие-либо процессы. Максимальная энтропия системы соответствует состоянию равновесия. Когда такое состояние достигается, то любые изменения в системе без внешних воздействий прекращаются. Любой самопроизвольный процесс в таком состоянии приводил бы к уменьшению энтропии, а, следовательно, к уменьшению вероятности такого состояния, что невозможно.
Поэтому состояние с максимальной энтропией – наиболее устойчивое состояние системы.
Энтропия может быть связана с величиной термодинамической вероятности, поскольку и та и другая величина имеет максимальное значение в состоянии равновесия и переход всякой системы в состояние равновесия сопровождается как возрастанием энтропии, так и термодинамической вероятности.
Статистическая причина увеличения энтропии при протекании необратимых процессов заключается в том, что при этом увеличивается термодинамическая вероятность состояния системы. Поэтому энтропия S должна быть некоторой функцией термодинамической вероятности W. Больцман показал, что:
 (1)
(1)
где k – постоянная Больцмана, W – термодинамическая вероятность состояния, для которого подсчитывается энтропия.
Как было показано выше, состояние с большим беспорядком характеризуется большей термодинамической вероятностью, чем более упорядоченное состояние. Следовательно, чем больший беспорядок в системе, тем больше энтропия системы. Любой естественный процесс всегда протекает так, что система переходит в состояние с большим беспорядком: температуры разных тел выравниваются, газы перемешиваются и т.д. Поэтому энтропию можно считать мерой беспорядка системы.
С беспорядком связана и необратимость тепловых процессов: тепловые процессы всегда протекают так, что беспорядок в системе увеличивается. С этим связан и тот факт, что любой вид энергии, в конце концов, переходит в теплоту, потому что тепловая энергия это энергия беспорядочного, хаотичного движения молекул, а все другие виды энергии связаны с более упорядоченным движением.
Связь между энтропией и вероятностью позволяет несколько иначе трактовать II начало термодинамики. II начало термодинамики утрачивает свою категоричность: если система находится в каком-либо состоянии с данным значением энтропии, то с очень большой вероятностью она перейдет в состояние с большей энтропией.
Это означает, что более вероятным изменением энтропии является ее рост. Однако, принципиально возможны и процессы, сопровождающиеся и уменьшением энтропии. Например, флуктуации давления газа, его плотности это такие изменения состояния, которые сопровождаются уменьшением энтропии и, следовательно, вероятности данного состояния. Но эти малые отклонения от равновесного состояния не противоречат II началу термодинамики, они являются следствием именно вероятностного характера энтропии.
Уравнение:
 (2)
(2)
позволяет определить не абсолютное значение энтропии, а только разность ее значений в двух состояниях. Определить значение энтропии в любом состоянии дает возможность теорема Нернста:
При абсолютном нуле температуры любые изменения состояния происходят без изменения энтропии.
Либо:
При стремлении температуры к абсолютному нулю энтропия любого тела (системы) также стремится к нулю.
 (3)
(3)
Согласно этой теореме энтропия любого тела (системы) равна нулю, если T = 0, т.е.:
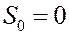
Следовательно, энтропия тела (системы) в состоянии с температурой T может быть рассчитана следующим образом:
 (4)
(4)
Для определения S из (6) необходимо знать молярную теплоемкость C, т.к. ее значение зависит от вида процесса и температуры.
Из теоремы Нернста следует, что абсолютный нуль температуры принципиально не достижим, т.к. при стремлении температуры к нулю, согласно (4) изменение энтропии должно стремиться к бесконечности, что противоречит теореме Нернста. Теорема Нернста применима только для систем, находящихся в состоянии термодинамического равновесия и не справедлива для неравновесных систем.
Сформулируем основные свойства энтропии:
1. Энтропия является функцией состояния системы;
2. Для подсчета изменения энтропии при переходе системы из состояния 1 в состояние 2 необходимо подсчитать значение интеграла (4) для любого обратимого процесса, приводящего систему из состояния 1 в состояние 2;
3. Энтропия замкнутой системы остается постоянной, если в системе протекают обратимые процессы и возрастает, если в системе протекают необратимые процессы;
4. Максимальное значение энтропии соответствует состоянию термодинамического равновесия системы;
5. Энтропия непосредственно связана с термодинамической вероятностью состояния системы;
6. Возрастание энтропии при необратимых процессах означает, что энергия, которой обладает система, становится менее доступной для преобразования в механическую работу;
7. В состоянии равновесия, когда энтропия максимальна, энергия системы вообще не может быть преобразована в работу.
Реальные газы и жидкости
Лекция 16
Отступление реальных газов от законов для идеальных газов. Взаимодействие молекул. Уравнение Ван-дер-Ваальса и его анализ. Критическое состояние. Экспериментальные изотермы реального газа. Сопоставление изотерм Ван-дер-Ваальса с экспериментальными изотермами.
Реальные газы при не слишком высоких давлениях и при не слишком низких температурах хорошо подчиняются газовым законам. При увеличении давления наблюдаются значительные отклонения от этих законов. При увеличении давления увеличивается плотность газа, при этом уменьшается среднее расстояние между молекулами. В таких условиях все большую роль начинает играть собственный объем молекул и взаимодействие между ними, что не учитывалось при получении уравнения Клапейрона-Менделеева и газовых законов.
Если учесть размеры молекул и силы притяжения между ними, то можно ввести соответствующие поправки в уравнение Клапейрона-Менделеева и получить уравнение состояния реального газа.
Объем, в котором могут двигаться молекулы меньше геометрического объема сосуда V на некоторую величину b, пропорциональную собственному объему молекул. Поправка b учитывает наличие сил отталкивания между молекулами.
Кроме сил отталкивания надо учитывать наличие сил притяжения между молекулами. Силы притяжения приводят к возникновению добавочного давления ∆ p, которое называется молекулярным давлением.
С учетом поправок на силы притяжения и отталкивания уравнение состояния для 1 моля реального газа примет вид:
 (1)
(1)
где Vµ - объем 1 моля газа.
Определим величину молекулярного давления ∆ p. Рассмотрим молекулы, которые находятся у стенки сосуда. На эти молекулы действует сила притяжения со стороны молекул, находящихся вдали от стенки. Эта сила направлена внутрь сосуда, и она уменьшает давление газа на стенку. Число внутренних молекул, а значит и сила, которая действует со стороны их, на каждую пристеночную молекулу пропорциональна числу молекул в единице объема n. Число молекул, находящихся у стенки сосуда также пропорционально концентрации n. Следовательно, сила, с которой внутренние молекулы действуют на пристеночные молекулы, пропорциональна n 2:

Известно, что:
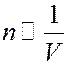
Значит величина силы, действующей на пристеночные молекулы:

Эта сила уменьшает давление на стенку сосуда на величину ∆ p:

Или:
 (2)
(2)
где a – коэффициент пропорциональности, зависящий от рода газа.
С учетом (2) из (1) получим уравнение состояния 1 моля реального газа:
 (3)
(3)
где Vµ - объем 1 моля газа.
Уравнение (3) получил в 1873 году голландский физик Ван-дер-Ваальс, и оно носит его имя.
Учтем, что:
 (4)
(4)
где V – объем ν молей газа, ν = m / µ - число молей.
С учетом (4) из (3) получим уравнение Ван-дер-Ваальса для произвольной массы газа:
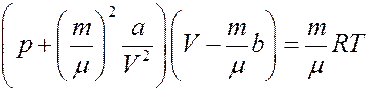 (5)
(5)
Постоянные a и b для каждого газа определяются опытным путем. Уравнение (5) значительно лучше описывает состояние реального газа, чем уравнение Клапейрона-Менделеева.
Определим величину поправки b. Две молекулы могут сблизиться на расстояние не меньше, чем сумма их радиусов (рис.1).

Рис.1. К выводу значения поправки на объем молекул
Очевидно, что из всего объема, который занимает газ необходимо исключить некоторую часть b, пропорциональную объему сферы с радиусом 2 r и числу сталкивающихся молекул:

где:
 ,
,
собственный объем молекулы, n – концентрация молекул, α - коэффициент, учитывающий число сталкивающихся молекул. Он равен ½, т.к. в большинстве случаев одновременно сталкивается две молекулы. С учетом этого:

где Vc – собственный объем молекул в единице объема.
Преобразуем уравнение (3). Обозначим объем 1 моля газа просто V и раскроем скобки:

Умножим обе части на V 2:

Разделим обе части на p и перегруппируем члены:
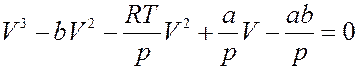
Или:
 (6)
(6)
Уравнение третей степени относительно V имеет либо три вещественных корня, либо один. Каждому корню на плоскости p, V соответствует точка, в которой изобара (p = const) пересекает изотерму Ван-дер-Ваальса. Если уравнение (6) имеет три корня, то каждому значению давления p и температуры T соответствует три значения объема V 1, V 2, и V 3 (рис.2).

Рис.2. Изотерма Ван-дер-Ваальса
Теоретические кривые, которые соответствуют уравнению (6), построенные при разных температурах называются изотермами Ван-дер-Ваальса (рис.3).

Рис.3. Изотермы Ван-дер-Ваальса. T 1< T 2< T к< T 3< T 4.
При высоких температурах T 3, T 4 изотермы Ван-дер-Ваальса практически не отличаются от изотерм идеального газа. При понижении температуры на изотермах появляются изгибы, которые увеличиваются при дальнейшем понижении температуры (рис.3). В этих областях температур каждому значению давления соответствует три значения объема. Это значит, что все три корня уравнения (6) вещественные.
При повышении температуры точки V 1, V 2, V 3, которые соответствуют решениям (6) сближаются и при некоторой температуре, разной для различных веществ, сливаются в одну точку. Это значит, что корни уравнения (6) становятся кратными и совпадают. Эта точка называется критической точкой (К). Соответствующие этой точке значения давления, объема и температуры называются критическими (p к, V к, T к) (рис.3).
Значения p к, V к, T к разные для разных веществ. Их можно однозначно определить через постоянные a и b уравнения Ван-дер-Ваальса. Для критического состояния все корни (6) равны:

Следовательно, должно выполняться равенство:

Или:
 (7)
(7)
Уравнение (7) должно быть тождественно уравнению (6) если в нем p = p к и T = T к:
 (8)
(8)
(7) и (8) тождественны, если коэффициенты при V равные:
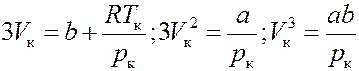 (9)
(9)
Разделим третье уравнение в (9) на второе:

Подставим V к во второе уравнение (9):

Подставим p к и V к в первое уравнение (9):

Таким образом, критические параметры определяются выражениями:
 (10)
(10)
Изотермы для одной и той же температуры будут разные для разных газов, т.к. постоянные a и b и связанные с ними критические параметры разные для разных газов. Используя приведенные параметры можно получить уравнение Ван-дер-Ваальса не зависящее от рода газа. Приведенными параметрами называются отношения:
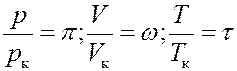
Отсюда следует:

Или:
 (11)
(11)
Подставив (11) в уравнение Ван-дер-Ваальса:
 ,
,
получим:
 (12)
(12)
Из (12) следует, что если разные вещества находятся в состоянии с двумя одинаковыми приведенными параметрами из трех, то и третий параметр будет также одинаковый.
При температурах ниже критической между теоретическими изотермами Ван-дер-Ваальса и изотермами, полученными экспериментально, наблюдаются значительные расхождения. Первые экспериментальные изотермы были получены Эндрюсом в 1866 году при изотермическом сжатии углекислого газа (рис.4).

Рис.4. Изотермы реального газа
При высоких температурах экспериментальные и теоретические изотермы практически одинаковые. При низких температурах уменьшение объема сначала также сопровождается ростом давления. При уменьшении объема до некоторого значения V 2 давление перестает увеличиваться, несмотря на уменьшение объема (точка a). При этом сжимаемый газ перестает быть однородным, часть его конденсируется в жидкость. При дальнейшем уменьшении объема все большая часть газа переходит в жидкое состояние, давление при этом не изменяется – линия a - c (рис. 4). При уменьшении объема до V 1 газ полностью переходит в жидкое состояние (точка c). Дальнейшее уменьшение объема сопровождается резким увеличением давления, что обусловлено очень малой сжимаемостью жидкости.
Таким образом, вместо S – образного участка теоретической изотермы Ван-дер-Ваальса, экспериментальные изотермы имеют горизонтальный участок a – c, который соответствует разделению вещества на две фазы: жидкую и газообразную. На этом участке имеет место равновесие между жидкой и газообразной фазами вещества.
Газ или пар, находящийся в равновесии со своей жидкостью называется насыщенным паром. Давление при данной температуре, при котором пар становится насыщенным, называется давлением или упругостью насыщенного пара. Величина давления насыщенного пара зависит только от температуры и не зависит от занимаемого объема.
При повышении температуры горизонтальные участки (изобары насыщенного пара), которые соответствуют двухфазному состоянию, уменьшаются. Это происходит потому, что с ростом температуры удельный объем насыщенного пара уменьшается, точка a сдвигается влево, а удельный объем жидкости увеличивается, точка c сдвигается вправо, (плотность жидкости уменьшается, а плотность пара увеличивается).
Удельным объемом называется объем единицы массы веществ а:

При некоторой температуре (31,1oC для CO2) точки a и c сливаются в точку К, при этом исчезает разница между жидким и газообразным состоянием вещества.
При некоторых, специальных условиях можно получить состояния, соответствующие участкам c - d и a - f теоретических изотерм Ван-дер-Ваальса (рис.5):

Рис.5. Теоретическая и экспериментальная изотермы реального газа
Например, участок c - d можно получить при сжатии газа, или при резком охлаждении сосуда с воздухом, содержащим пары воды. При этом газ (воздух) должен быть очищен от пыли, ионов и т.д. (центры конденсации). При этих условиях, несмотря на охлаждение и уменьшение, таким образом, упругости насыщенного пара воды, пар может не конденсироваться некоторое время, оставаясь в газообразном состоянии. Такой пар называется пересыщенным. Однако такое состояние неустойчивое. В конце концов, конденсация начинается, давление скачком уменьшается до давления насыщенного пара при данной температуре.
При изотермическом расширении жидкости можно получить задержку в появлении пара – участок a – f, несмотря на то, что давление пара будет меньше, чем давление насыщенного пара при данной температуре. Это состояние неустойчивое (метастабильное). Оно называется состоянием растянутой или перегретой жидкости. Перегретой в смысле того, что при данных значениях температуры и объема часть жидкости должна находиться в парообразном состоянии, в то время как пар отсутствует. Состояния, которые соответствуют участкам f - b - d изотермы Ван-дер-Ваальса, вообще не могут существовать. При этих условиях вещество должно обладать неестественными свойствами: при уменьшении объема, давление должно уменьшаться и наоборот.
При некоторой температуре T к горизонтальная линия, которая соответствует двухфазной системе, стягивается в точку (точка перегиба кривой). При этой температуре, разной для разных веществ, исчезает разница между жидким и газообразным состояниями. Это состояние называется критическим состоянием. Критическая температура гелия – 4K, воды – 374оC. В критической точке каждое вещество характеризуется критическими параметрами. Например, для воды: T к = 374 оC, p к = 218 атм., V к = 3см3 для 1 грамма воды.
В критическом состоянии переход вещества из жидкого состояния в газообразное и наоборот осуществляется беспрерывно. Вблизи критического состояния вещество в газообразном состоянии становится мутным, благодаря интенсивному рассеянию света на неоднородностях среды. Это явление называется критической опалесценцией. Вблизи критического состояния возникают флуктуации плотности газа. Точка К – точка перегиба кривой, касательная к кривой в этой точке горизонтальная (рис.4). Поэтому вблизи точки К dp / dV ≈ 0. Следовательно, вблизи критической точки значительные изменения плотности вещества (dV) вызывают незначительное изменение давления (dp). Поэтому флуктуации плотности могут быть значительными и приводить к возникновению мельчайших капелек жидкости, на которых и рассеивается свет.
Вдали от критического состояния dp / dV ≠ 0 и, поэтому изменения плотности приводят к значительному изменению давления, которое быстро выравнивается. Следовательно, вдали от критического состояния значительных флуктуаций плотности наблюдаться не будет.
Если соединить между собой крайние точки a и c горизонтальных участков семейства экспериментальных изотерм, то получится дугообразная кривая, которая разделяет pV диаграмму на три области (рис.6):

Рис.6. Изотермы реального газа. Область I – жидкость, область II – двухфазная система «жидкость + насыщенный пар», область III – газообразное вещество. K – критическая точка.
Если вещество находится в области III при T > T к, то никаким сжатием оно не может быть переведено в жидкое состояние.
Выбрав процесс перехода, чтобы он нигде не пересекал двухфазную область II, можно осуществить переход из жидкого состояния в газообразное (A → B → C) и наоборот (C → B → A) без разделения вещества на две фазы. В этом случае, в процессе перехода вещество все время будет оставаться однородным.
Лекция 17