

Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
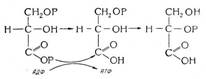
Биохимия спиртового брожения: Основу технологии получения пива составляет спиртовое брожение, - при котором сахар превращается...
Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Биохимия спиртового брожения: Основу технологии получения пива составляет спиртовое брожение, - при котором сахар превращается...
Топ:
Когда производится ограждение поезда, остановившегося на перегоне: Во всех случаях немедленно должно быть ограждено место препятствия для движения поездов на смежном пути двухпутного...
Установка замедленного коксования: Чем выше температура и ниже давление, тем место разрыва углеродной цепи всё больше смещается к её концу и значительно возрастает...
Интересное:
Как мы говорим и как мы слушаем: общение можно сравнить с огромным зонтиком, под которым скрыто все...
Мероприятия для защиты от морозного пучения грунтов: Инженерная защита от морозного (криогенного) пучения грунтов необходима для легких малоэтажных зданий и других сооружений...
Искусственное повышение поверхности территории: Варианты искусственного повышения поверхности территории необходимо выбирать на основе анализа следующих характеристик защищаемой территории...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Дата __________
______________ _______________ ______________________________
ДолжностьподписьФ.И.О. ответственного лица
Заявителя
Место печати
Приложение 2
к Правилам государственной регистрации,
перерегистрации и внесения изменений
в регистрационное досье лекарственных средств
Форма
Заявление
на внесение изменений в регистрационное досье лекарственного
средства, прошедшего государственную регистрацию
(перерегистрацию) в Республике Казахстан
| № _______ | «____» ________ 200_ г. |
1. Торговое название лекарственного средства (на государственном, русском языках) ___________________________________________
2. Лекарственная форма, дозировка, концентрация, объем заполнения, количество доз в упаковке_________________________________
3. Лекарственное средство зарегистрировано в Республике Казахстан и внесено в Государственный реестр под №_________ от_______
4. Заявитель (организация-производитель, владелец регистрационного удостоверения, доверенное лицо) (нужное заполнить):
4.1. Организация-производитель лекарственного средства Наименование организации-производителя ___________________________
Страна организации-производителя (полное наименование страны (официальный статус) с указанием цифрового кода по единому классификатору ГК РК ИСО 3166)
Юридический адрес __________________________________________________________________________________________________
Адрес местонахождения_______________________________________________________________________________________________
Фамилия, имя руководителя ___________________________________________________________________________________________
Телефон, факс, Е-mail__________________________________________________________________________________________________
№ договора на проведение экспертизы лекарственных средств, дата заключения, срок действия __________________________________
|
|
Владелец регистрационного удостоверения
Название компании _________________________________________________________________________________________________
Предприятие-производитель _________________________________________________________________________________________
(указать для заполнения регистрационного удостоверения)
на государственном языке _____________________________________________________________________________________________
на русском языке _____________________________________________________________________________________________________
для зарубежных (кроме стран СНГ) (дополнительно на английском языке)_____________________________________________________
Страна предприятия-производителя (полное наименование страны (официальный статус) с указанием цифрового кода по единому
классификатору ГК РК ИСО 3166) ______________________________________________________________________________________
Руководитель ________________________________________________________________________________________________________
Юридический адрес___________________________________________________________________________________________________
Адрес местонахождения _______________________________________________________________________________________________
Телефон, факс, Е-mail __________________________________________________________________________________________________
№ договора на проведение экспертизы лекарственных средств, дата
заключения, срок действия ____________________________________________________________________________________________
Доверенное лицо/компания, представительство от заявителя, уполномоченное проводить действия во время процедуры
государственной регистрации в Республике Казахстан __________________________________________________________________
действующее на основании ____________________________________________________________________________________________
(доверенность №, дата выдачи, срок действия)
Фамилия, имя, отчество/название компании______________________________________________________________________________
Юридический адрес __________________________________________________________________________________________________
Адрес местонахождения _______________________________________________________________________________________________
Телефон, факс, E-mail __________________________________________________________________________________________________
№ договора на проведение экспертизы лекарственных средств, дата
заключения, срок действия ____________________________________________________________________________________________;
|
|
4.4. Доверенное лицо/компания, представительство от заявителя, уполномоченное проводить действия между владельцем регистрационного удостоверения и уполномоченным органом Республики Казахстан после государственной регистрации (если они иные, чем определенные подпунктом 4.3. ) _________________________________________________________________________________
действующее на основании ____________________________________________________________________________________________
(доверенность №, дата выдачи, срок действия)
Фамилия, имя, отчество/название компании ______________________________________________________________________________
Юридический адрес___________________________________________________________________________________________________
Адрес местонахождения _______________________________________________________________________________________________
Телефон, факс, E-mail __________________________________________________________________________________________________
№ договора на проведение экспертизы лекарственных средств, дата
заключения, срок действия ____________________________________________________________________________________________;
5. Изменения, которые заявляются: ______________________________________________________________________________________
____________________________________________________________________________________________________________________
____________________________________________________________________________________________________________________
____________________________________________________________________________________________________________________
____________________________________________________________________________________________________________________
Заявленные данные:
Том _____________ страницы ________________
6. Качественный и количественный состав лекарственного средства, включая действующие и вспомогательные вещества
| Вещество | Количество на единицу лекарственной формы |
| Активные вещества | |
| 1. | |
| 2. | |
| 3. и т.д. | |
| Вспомогательные вещества | |
| 1. | |
| 2. и т.д. |
Заявитель гарантирует достоверность информации, которая представлена, и сохраняет ответственность за эффективность, безопасность и качество лекарственного средства.
Плательщик экспертизы лекарственного средства является__________________________________________________________________
____________________________________________________________________________________________________________________
(организация-производитель, представительство, доверенное лицо)
Реквизиты плательщика: _______________________________________________________________________________________________
(РНН, ИИН, БИН, р/с, в/с, код, БИК, банк)
Дата __________
_____________ ________________ __________________________________
Должность подпись Ф.И.О. руководителя фирмы или
официального представителя
Место печати
|
|
Приложение 3
к Правилам государственной регистрации,
перерегистрации и внесения изменений
в регистрационное досье лекарственных
средств
Перечень
документов регистрационного досье, предоставляемых при
государственной регистрации, перерегистрации лекарственных
средств в Республике Казахстан
| № п/п | Наименование документов | Лекарственные средства (ЛС) | ||||||
| Лекарственный препарат | Лекарственный бал-кпродукт | Лекарственная субстанция | Нефармакопейное лекарственное растительное сырье | Гомео- патические лекарственные препараты | Пара фармацевтики | Медицинские иммунобио- логические препараты | ||
| Часть I Общая документация | ||||||||
| I А | Административные данные | |||||||
| I А 1. | Заявление на государственную регистрацию по форме (на бумажном и электронном носителях) | + | + | + | + | + | + | + |
| I А 2. | **Сертификат на фармацевтический продукт (СРР), выданный согласно рекомендации ВОЗ | + | + | - | + | + | + | + |
| При отсутствии предоставляются: | ||||||||
| **Сертификат (регистрационное удостоверение) о регистрации в стране-производителе (заверенные нотариально) | + | + | - | - | + | + | + | |
| **Сертификат GMP c указанием даты и результатов последней инспекции) (заверенные нотариально) | + | + | - | - | + | + | + | |
| Сертификат, разрешающий свободную продажу (экспорт) | + | + | - | + | + | + | + | |
| I А 3. | ***Государственную лицензию на фармацевтическую деятельность (заверенную нотариально) | + | + | + | + | + | + | + |
| I А 4. | ***Приложение к лицензии (для растительного сырья - разрешение на заготовку для отечественных производителей) | + | + | + | + | + | + | + |
| I А 5. | Если в производственном процессе участвует несколько производителей документы пунктов IА2, IА3, IА4 предоставляются на всех участников производства | + | + | + | + | + | + | + |
| I А 6. | Лицензионный договор (соглашение) на право производства (до истечения срока действия патента на оригинальный препарат) | + | + | - | - | + | - | + |
| I А 7. | Сведения о регистрации в других странах с указанием номера и даты регистрационного удостоверения (или копии сертификата или регистрационного удостоверения) | + | + | + | + | + | + | + |
| I А 8. | Документ, подтверждающий качество активного вещества (сертификат анализа субстанции от производителя, сертификат соответствия монографии Европейской Фармакопеи, протокол анализа, аналитический паспорт и др.) | + | + | - | - | - | + | + |
| I А 9. | Документ, подтверждающий качество готового продукта трех промышленных серий (сертификат анализа, протокол анализа и др.), одна серия которого должна совпадать с серией образца ЛС, поданного на регистрацию | + | + | - | + | + | + | + |
| I А 10. | Документ о прионовой безопасности на вещества животного происхождения от производителя | + | + | + | - | + | + | + |
| I А 11. | Копия - регистрационного удостоверения РК при перерегистрации | + | + | + | + | + | + | + |
| I А 12. | Сведения об отказе в регистрации, отзыве с рынка компетентным органом или заявителем, о прекращении действия регистрационного удостоверения или приостановлении его компетентным органом (с указанием причины в случае имеющихся прецедентов) | + | + | + | + | + | + | + |
| I.В. | Краткая характеристика лекарственного препарата (SPC), маркировка, инструкция по медицинскому применению | |||||||
| I.В.1. | ** Краткая характеристика лекарственного препарата (SPC), утвержденная в стране-производителе на английском языке | + | - | - | + | + | + | + |
| I.В.2. | **Аутентичный перевод краткой характеристики лекарственного препарата (SPC) на русский язык | + | - | - | + | + | + | + |
| I.В.3. | Проект инструкции по медицинскому применению на государственном языке (бумажном и электронном носителях) | + | - | - | + | + | + | + |
| I.В.4. | Проект инструкции по медицинскому применению на русском языке (бумажном и электронном носителях) | + | - | - | + | + | + | + |
| I.В.5. | Цветные макеты упаковок и этикеток на электронном и бумажном носителях (при отсутствии образцов торговых упаковок – экземпляр в конечной первичной упаковке, без конечной маркировки. При этом экземпляр в конечной первичной и вторичной упаковках должен быть представлен дополнительно до окончания проведения экспертизы) в 3-х экземплярах | + | - | - | + | + | + | + |
| I.С | Детальное описание фармаконадзора и системы управления риском при медицинском применении лекарственного препарата, предлагаемой заявителем | + | - | - | - | + | + | + |
| I.D | Документ, подтверждающий о наличии квалифицированного лица, ответственного за фармаконадзор для сбора и регистрации побочных реакций, выявляемых на территории РК | + | - | - | - | + | + | + |
| Часть II Химическая, фармацевтическая и биологическая документация | ||||||||
| II | Содержание | + | + | + | + | + | + | + |
| II А | Состав | |||||||
| II А 1 | Качественный и количественный состав лекарственного препарата (активные, вспомогательные вещества, состав оболочки таблетки или корпуса капсулы) | + | + | - | + | + | + | + |
| II А 2 | Упаковка (краткое описание) | + | + | - | + | + | + | + |
| II А 3 | Фармацевтическая разработка (обоснование выбора состава, первичной упаковки и др.) | + | + | - | + | + | + | + |
| II В | Сведения о производстве: | |||||||
| II В 1 | производственная формула | + | + | - | - | - | + | + |
| II В 2 | описание технологии производства | + | + | + (путь синтеза) | - | + | + | + |
| II В 3 | контроль в процессе производства (операционный контроль) | + | + | - | - | + | + | + |
| II В 4 | валидация производственных процессов | + | + | - | - | + | + | + |
| II С | Методы контроля исходных материалов | |||||||
| II С 1 | Активная субстанция | |||||||
| II С 1.1 | сертификаты качества на действующие вещества (кроме фармакопейных) | + | + | - | + | + | + | + |
| II С 2 | Вспомогательные вещества | |||||||
| II С 2.1 | сертификаты качества на вспомогательные вещества | + | + | - | - | + | + | + |
| II С 3 | Упаковочный материал (первичная и вторичная упаковка) | |||||||
| II С 3.1 | сертификаты качества на упаковочный материал | + | + | + | + | + | + | + |
| II E | Спецификация качества готового продукта с аутентичным переводом | |||||||
| II E 1 | Утвержденный нормативно-технический документ по контролю за качеством и безопасностью ЛС, пояснительная записка к нему, валидация методик испытаний лекарственного препарата (кроме фармакопейных методик) на бумажном и электронном носителях | + | + | + | + | + | + | + |
| II F | Результаты испытания стабильности не менее чем на 3-х промышленных или опытно-промышленных (пилотных) сериях | + | + | + | + | + | + | + |
| II G | Сведения о профиле растворения (для твердых дозированных лекарственных форм) | + | + | - | - | + | + | |
| II H | Данные по биодоступности, биоэквивалентности (для генериков), для парентеральных форм генериков - данные по безопасности и эффективности | + | - | - | - | - | + | |
| II K | Данные контроля на животных | - | - | - | - | - | - | + |
| II L | Данные по вероятной опасности для окружающей среды для препаратов, содержащих генетически измененные организмы | + | + | + | - | - | + | + |
| II M | Периодический обновляемый отчет по безопасности (при перерегистрации) | + | - | - | + | + | - | + |
| II Q. | Дополнительная информация, подтверждающая качество (при необходимости) | + | + | + | + | + | + | + |
| Часть III. Фармакологическая и токсикологическая документация | ||||||||
| III. | Содержание | + | + | - | + | + | + | + |
| III А. | Данные по токсичности (острой и хронической), (МИБП - токсичность при однократном введении и введении повторных доз) | + | - | - | - | + | + | + |
| III В. | Влияние на репродуктивную функцию | + | - | - | - | - | + | + |
| III С. | Данные по эмбриотоксичности и тератогенности | + | - | - | - | - | + | + |
| III D. | Данные по мутагенности | + | - | - | - | - | + | + |
| III Е. | Данные по канцерогенности | + | - | - | - | - | + | + |
| III F. | Фармакодинамика (МИБП - реактогенность) | + | - | - | - | + | + | + |
| III G. | Фармакокинетика (МИБП - специфическая активность) | + | - | - | - | - | + | + |
| III H. | Данные о местно- раздражающем действии (МИБП - иммуногенность для вакцин) | + | - | - | - | - | + | + |
| III Q. | Дополнительная информация, подтверждающая безопасность (при необходимости) | + | - | - | - | + | + | + |
| Часть IV. Клиническая документация | ||||||||
| IV. | Содержание | + | - | - | - | + | + | |
| IV А. | Данные по клинической фармакологии (фармакодинамика, фармакокинетика) | + | - | - | - | + | + | + |
| IV В | Клиническая, иммунологическая эффективность | - | - | - | - | - | - | + |
| IV С | Диагностическая эффективность | - | - | - | - | - | - | - |
| IV D | Результаты клинических испытаний, научные публикации, отчеты | + | - | - | - | + | + | + |
| IV D 1 | Данные пострегистрационного опыта (при наличии) | + | - | - | - | + | + | + |
| IV Q | Дополнительная информация, подтверждающая эффективность | + | - | - | - | + | + | + |
|
|
|
|
Приложение к регистрационному досье (заполняется в двух экземплярах):
| Наименование, лекарственная форма, дозировка, концентрация, объем, количество доз в упаковке | Ед. изм | Кол- во | Условия хранения | |
| 1. | Образцы лекарственного средства в упаковке в количестве, достаточном для проведения 3-х кратного анализа | |||
| 2. | Стандартные образцы для определения посторонних примесей (при необходимости) | |||
| 3. | Образцы субстанции для проведения 3-х кратного анализа | |||
| 4. | Стандартные образцы действующего вещества для анализа субстанции | |||
| 5. | Расходные материалы (в исключительных случаях и на условиях возврата) |
Сдал (Ф.И.О.) _________________ Подпись _________
Принял (Ф.И.О.) _______________ Подпись _________
Дата
Примечание:
** - документы предоставляются только организациями-производителями дальнего зарубежья;
*** - документы предоставляются только организациями-производителями стран СНГ и Республики Казахстан;
документы, не имеющие обозначения, обязательны для всех заявителей.
Приложение 4
к Правилам государственной регистрации,
перерегистрации и внесения изменений
в регистрационное досье лекарственных средств
Перечень
документов регистрационного досье, предоставляемых при государственной регистрации, перерегистрации лекарственных
средств в Республике Казахстан в формате Общего технического документа
(для лекарственных средств, произведенных в условиях Надлежащей производственной практики)
| № п/п | Наименование документов |
| Модуль 1. | |
| Административная информация: | |
| 1.1. | Общая документация |
| 1.2 | Заявление на государственную регистрацию по форме (на бумажном и электронном носителях) |
| 1.2.1 | Сертификат на фармацевтический продукт согласно рекомендации ВОЗ. При отсутствии предоставляются: Сертификат (регистрационное удостоверение) о регистрации в стране-производителе (заверенные нотариально) |
| 1.2.2. | Сертификат GMP (ВОЗ) (с указанием даты и результатов последней инспекции) (заверенные нотариально) |
| 1.2.3. | Сертификат, разрешающий свободную продажу (экспорт) |
| 1.2.4. | Сертификат происхождения товара (для отечественных производителей) |
| 1.2.5. | Лицензионный договор (соглашение) на право производства (до истечения срока действия патента на оригинальный препарат) |
| 1.2.6. | Сведения о регистрации ЛС в других странах с указанием номера и даты регистрационного удостоверения (или копии сертификата или регистрационного удостоверения) |
| 1.3. | Краткая характеристика лекарственного препарата, маркировка и инструкция по медицинскому применению |
| 1.3.1. | Краткая характеристика лекарственного препарата, утвержденная в стране производителя |
| 1.3.2. | Маркировка |
| 1.3.3. | Инструкция по медицинскому применению |
| 1.3.4. | Цветные макеты потребительской упаковки (при отсутствия образцов торговых упаковок – экземпляр в конечной первичной упаковке без конечной маркировки. При этом экземпляр в конечной первичной и вторичной упаковках должен быть представлен дополнительно до окончания экспертизы на бумажном и электронном носителях в масштабе 1:1 |
| 1.4. | Информация об экспертах |
| 1.4.1. | Информация об эксперте по качеству |
| 1.4.2. | Информация об эксперте по доклиническим данным |
| 1.4.3. | Информация об эксперте по клиническим данным |
| 1.5. | Специальные требования к разным типам заявлений |
| 1.5.1. | Информация относительно библиографических заявлений в соответствии со статьей 4.8 (ii) Директивы 65/65/ЕЕС |
| 1.5.2. | Информация относительно сокращенных заявлений в соответствии со статьей 4.8 (iii) Директивы 65/65/ЕЕС, 1 и 2 абзацы |
| 1.6. | Оценка потенциальной опасности для окружающей среды (Приложение 1 к модулю) |
| 1.6.1 | Лекарственные препараты, содержащие или полученные из геномодифицированных организмов |
| 1.7. | Информация относительно фармаконадзора заявителя в РК |
| 1.7.1 | Детальное описание фармаконадзора и системы управления риском при медицинском применении лекарственного препарата, предлагаемой заявителем |
| 1.7.2 | Документ, подтверждающий, что заявитель имеет в своем распоряжении квалифицированное лицо, ответственное за фармаконадзор на территории Республики Казахстан |
| Модуль 2. | |
| Резюме ОТД | |
| 2.1. | Содержание модулей 2.3.4.5 |
| 2.2. | Введение в ОТД |
| 2.3. | Общий отчет по качеству |
| 2.4. | Обзор доклинических данных |
| 2.5. | Обзор клинических данных |
| 2.6. | Отчет доклинических данных |
| 2.6.1. | Отчет фармакологических данных в текстовом формате |
| 2.6.2. | Отчет фармакологических данных в виде таблиц |
| 2.6.3. | Отчет фармакокинетических данных в текстовом формате |
| 2.6.4. | Отчет фармакокинетических данных в виде таблиц |
| 2.6.5. | Отчет оксикологических данных в текстовом формате |
| 2.6.6. | Отчет токсикологических данных в виде таблиц |
| 2.7. | Отчет клинических данных |
| 2.7.1. | Отчет биофармацевтических исследований и связанных с ними аналитических методов |
| 2.7.2. | Отчет исследований по клинической фармакологии |
| 2.7.3. | Отчет по клинической эффективности |
| 2.7.4. | Отчет по клинической безопасности |
| 2.7.5. | Копия использованных литературных источников |
| 2.7.6. | Короткие обзоры индивидуальных испытаний |
| Модуль 3. Качество | |
| 3.1. | Содержание |
| 3.2. | Основные данные |
| 3.2.S. | Лекарственное вещество (для лекарственных препаратов, которые содержат более одного лекарственного вещества, информация предоставляется в полном объеме относительно каждого из них)* |
| 3.2.S.1. | Общая информация* |
| 3.2.S.1.1. | Название* |
| 3.2.S.1.2. | Структура* |
| 3.2.S.1.3. | Общие свойства* |
| 3.2.S.2. | Производство* |
| 3.2.S.2.1. | Производитель* |
| 3.2.S.2.2. | Описание производственного процесса и его контроль* |
| 3.2.S.2.3. | Контроль материалов |
| 3.2.S.2.4. | Контроль критических этапов и промежуточной продукции |
| 3.2.S.2.5. | Валидация процесса и/или его оценка |
| 3.2.S.2.6. | Разработка производственного процесса |
| 3.2.S.3. | Характеристика* |
| 3.2.S.3.1. | Доказательство структуры и другие характеристики |
| 3.2.S.3.2. | Примеси* |
| 3.2.S.4. | Контроль лекарственного вещества* |
| 3.2.S.4.1. | Спецификация* |
| 3.2.S.4.2. | Аналитические методики* |
| 3.2.S.4.3. | Валидация аналитических методик |
| 3.2.S.4.4. | Анализы серий* |
| 3.2.S.4.5. | Обоснование спецификации |
| 3.2.S.5. | Стандартные образцы или вещества |
| 3.2.S.6. | Система упаковка/укупорка* |
| 3.2.S.7. | Стабильность* |
| 3.2.S.7.1. | Резюме относительно стабильности и выводы* |
| 3.2.S.7.2. | Протокол пострегистрационного изучения стабильности и обязательства относительно стабильности* |
| 3.2.S.7.3. | Данные о стабильности* |
| 3.2.Р. | Лекарственный препарат |
| 3.2.Р.1. | Описание и состав лекарственного препарата |
| 3.2.Р.2. | Фармацевтическая разработка |
| 3.2.Р.2.1. | Составные вещества лекарственного препарата |
| 3.2.Р.2.1.1. | Лекарственная субстанция |
| 3.2.Р.2.1.2. | Вспомогательные вещества |
| 3.2.Р.2.2. | Лекарственный препарат |
| 3.2.Р.2.2.1. | Разработка состава |
| 3.2.Р.2.2.2. | Излишки |
| 3.2.Р.2.2.3. | Физико-химические и биологические свойства |
| 3.2.Р.2.3. | Разработка производственного процесса |
| 3.2.Р.2.4. | Система упаковка/укупорка |
| 3.2.Р.2.5. | Микробиологические характеристики |
| 3.2.Р.2.6. | Совместимость |
| 3.2.Р.3. | Производство |
| 3.2.Р.3.1. | Производитель (и) |
| 3.2.Р.3.2. | Состав на серию |
| 3.2.Р.3.3. | Описание производственного процесса и контроля процесса |
| 3.2.Р.3.4. | Контроль критических этапов и промежуточной продукции |
| 3.2.Р.3.5. | Валидация процесса и/или его оценка |
| 3.2.Р.4. | Контроль вспомогательных веществ |
| 3.2.Р.4.1. | Спецификации |
| 3.2.Р.4.2. | Аналитические методики |
| 3.2.Р.4.3. | Валидация аналитических методик |
| 3.2.Р.4.4. | Обоснование спецификаций |
| 3.2.Р.4.5. | Вспомогательные вещества человеческого и животного происхождения |
| 3.2.Р.4.6. | Новые вспомогательные вещества |
| 3.2.Р.5. | Контроль лекарственного препарата |
| 3.2.Р.5.1. | Спецификация (и) |
| 3.2.Р.5.2. | Аналитические методики |
| 3.2.Р.5.3. | Валидация аналитических методик |
| 3.2.Р.5.4. | Анализы серий |
| 3.2.Р.5.5. | Характеристика примесей |
| 3.2.Р.5.6. | Обоснования спецификации (й) |
| 3.2.Р.6. | Стандартные образцы и вещества |
| 3.2.Р.7. | Система упаковка/укупорка |
| 3.2.Р.8. | Стабильность |
| 3.2.Р.8.1. | Резюме и вывод о стабильности |
| 3.2.Р.8.2. | Протокол пострегистрационного изучения стабильности и обязательства относительно стабильности |
| 3.2.Р.8.3. | Данные о стабильности |
| 3.2.А. | Дополнения |
| 3.2.А.1. | Технические средства и оборудование |
| 3.2.А.2. | Оценка безопасности относительно посторонних микроорганизмов |
| 3.2.А.3. | Новые вспомогательные вещества |
| 3.2.R. | Региональная информация |
| 3.3. | Копия использованных литературных источников |
| Модуль 4. Отчеты о доклинических (неклинических) исследованиях | |
| 4.1. | Содержание |
| 4.2. | Отчеты об исследовании |
| 4.2.1. | Фармакология |
| 4.2.1.1. | Первичная фармакодинамика |
| 4.2.1.2. | Вторичная фармакодинамика |
| 4.2.1.3. | Фармакология безопасности |
| 4.2.1.4. | Фармакодинамические лекарственные взаимодействия |
| 4.2.2. | Фармакокинетика |
| 4.2.2.1. | Аналитические методы и отчет относительно их валидации |
| 4.2.2.2. | Всасывание |
| 4.2.2.3. | Распределение |
| 4.2.2.4. | Метаболизм |
| 4.2.2.5. | Выведение |
| 4.2.2.6. | Фармакокинетические лекарственные взаимодействия (доклинические) |
| 4.2.2.7. | Другие фармакокинетические исследования |
| 4.2.3. | Токсикология |
| 4.2.3.1. | Токсичность при введении однократной дозы |
| 4.2.3.2. | Токсичность при введении повторных доз |
| 4.2.3.3. | Генотоксичность (invitro; invivo, токсикокинетическую оценку) |
| 4.2.3.4. | Канцерогенность (долгосрочные исследования; краткосрочные или среднерочные исследования) |
| 4.2.3.5. | Репродуктивная и онтогенетическая токсичность: способность к воспроизведению потомства и раннее эмбриональное развитие; развитие зародышаплода; внутриутробное и послеродовое развитие; исследования, в которых потомкам (растущие животные) давали определенную дозу и/или оценивали в дальнейшем. |
| 4.2.3.6. | Местная переносимость |
| 4.2.3.7. | Другие исследования токсичности: антигенность; иммунотоксичность; механические исследования; зависимость; метаболиты; примеси; другие. |
| 4.3. | Копия использованных литературных источников |
| Модуль 5. Отчеты о клинических исследованиях и (или) испытаниях | |
| 5.1. | Содержание |
| 5.2. | Перечень всех клинических испытаний в виде таблиц |
| 5.3. | Отчеты о клинических испытаниях |
| 5.3.1. | Отчеты о биофармацевтических исследованиях: отчет исследований по биодоступности; отчет сравнительных исследований по биодостпуности и биоэквивалентности; отчет по корреляции исследований invitroinvivo; отчет по биоаналитическим и аналитическим методам; |
| 5.3.2. | Отчеты исследований по фармакокинетике при использовании биоматериалов человека: отчет исследований связывания с белками; отчет исследований печеночного метаболизма и взаимодействий; отчет по исследованиям с использованием других биоматериалов человека. |
| 5.3.3. | Отчеты о фармакокинетических исследованиях у человека: отчет исследований фармакокинетики у здоровых добровольцев и исследованию первичной переносимости; отчет исследований фармакокинетики у пациентов и исследованию первичной переносимости; отчет исследований внутреннего фактора фармакокинетических исследований; отчет исследований внешнего фактора фармакокинетических исследований; отчет исследований фармакокинетики в различных популяциях; |
| 5.3.4. | Отчеты о фармакодинамических исследованиях у человека: отчет исследований фармакодинамики и фармакокинетики/фармакодинамики у здоровых добровольцев; отчет исследований фармакодинамики и фармакокинетики/фармакодинамики у пациентов; |
| 5.3.5. | Отчеты об исследовании эффективности и безопасности: отчет контролируемых клинических исследований по заявленным показаниям; отчет неконтролируемых клинических исследований; отчеты анализа данных более чем одного исследования, включая любые формальные интегрированные анализы, метаанализы и перекрестные анализы; отчеты по другим исследованиям. |
| 5.3.6. | Отчеты о пострегистрационном опыте применения |
| 5.3.7. | Образцы индивидуальных регистрационных форм и индивидуальные списки пациентов |
| 5.4. | Копия использованных литературных источников |
--------------------------------------------
* Минимальный объем сведений, который необходимо предоставлять в разделе 3.2.S.
Если отдельные части документации не включены в материалы, следует в соответствующим месте указать причину под соответствующим заглавием.
Для препаратов животного происхождения в разделе 3.2.S должна быть подана такая дополнительная информация:
данные относительно вида, возраста, рациона животных, от которых получено сырье;
данные о характере (категории) ткани, из которой получено сырье для производства лекарственного средства, с точки зрения его опасности относительно содержимого прионов;
технологическая схема обработки сырья с указанием экстрагентов, температурного режима и т.п.;
методы контроля исходного сырья, включая методы выявления прионов в конечном продукте (при необходимости).
_____________________________
Примечание. Приложение 4 составлено на основе документа: Rules governing medicinal product in the Europian Union, NTA, vol. 2B-CTD, 2001 (в случае использования других версий - приведите соответствующую ссылку).
Приложение 5
к Правилам государственной регистрации,
перерегистрации и внесения изменений
в регистрационное досье лекарственных средств
1. Изменения типа 1, вносимые в регистрационное досье
лекарственного средства
| Изменение | Условия/замечания | Перечень документов и материалов, необходимых для внесения изменений | |||
| 1. Изменение содержания Производственной лицензии: | Основное условие Новая лицензия на производство должна быть подана соответствующему органу | ||||
| Изменение названия производителя лекарственного средства | Место производства не измен<
 Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...  История развития пистолетов-пулеметов: Предпосылкой для возникновения пистолетов-пулеметов послужила давняя тенденция тяготения винтовок...  История развития хранилищ для нефти: Первые склады нефти появились в XVII веке. Они представляли собой землянные ямы-амбара глубиной 4…5 м...  Состав сооружений: решетки и песколовки: Решетки – это первое устройство в схеме очистных сооружений. Они представляют... © cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста. |