(при которых всегда возникает умственная отсталость).
I. Нарушение обмена аминокислот:
· Фенилкетонурия
· Гомоцистинурия
· Гистидиния
II. Нарушение обмена углеводов:
· Галактоземия (или галактозурия)
· Фруктозурия
III. Болезни накопления:
· Мукополисахаридозы
· Болезнь Гунтера (Гуллера)
· Липидозы
· Болезнь Нимана-Пика
· Амвротическая идиотия
· Заболевания, связанные с нарушениями эндокринной системы (эндермический зоб)
I. Нарушение обмена аминокислот:
Фенилкетонурия
Фенилкетонурия (ФКУ) – наследственное заболевание обмена, характеризующееся поражением ЦНС и прогрессирующим, особенно в первые 2–3 года жизни, слабоумием. Здоровые родители больного ребенка являются носителями мутантного гена. Частота заболевания в Европе в среднем составляет 1: 10 000 новорожденных. ФКУ наблюдается примерно у 1% умственно отсталых лиц
Заболевание обусловлено мутацией гена, контролирующего синтез фермента фенилаланингидроксилазы, который обеспечивает превращение поступающего в организм с пищей фенилаланина в тирозин
Дети с ФКУ рождаются с полноценным головным мозгом, так как биохимические процессы плода осуществляются за счет процессов в организме матери. Возникающие после рождения биохимические нарушения оказывают токсическое воздействие на нервную систему, в результате чего нарушаются процессы миелинизации, развитие и рост мозга.
Нарастание интеллектуального дефекта сочетается с отставанием в физическом развитии, часто с признаками умеренной микроцефалии. Характерен внешний вид больных (блондины со светлой кожей и голубыми глазами) и отдельные диспластические признаки (высокое небо, эпикант т.е.третье веко, деформация ушных раковин).
Уровень интеллектуального развития колеблется от нормы до глубокой идиотии.
Динамика слабоумия наиболее выражена в первые 2–3 года жизни. Больные отличаются инертностью, недостаточной целенаправленностью с характерными нарушениями внимания, памяти, недоразвитием гностических функций и пространственных представлений. Отмечается также выраженное недоразвитие речи и нарушения звукопроизношения. Нарушения речи обычно сопоставимы с глубиной интеллектуального дефекта.
Схема обмена фенилаланина и тирозина при фенилкетонурии

II. Нарушение обмена углеводов:
Галактоземия (или галактозурия)
Наследуется аутосомно-рецессивно (т.е. через поколение), частота больных — 1: 70 000 населения.
При этом заболевании – в организме: дефицит фермента, который должен расщеплять молочный сахар. При введении молока в связи с неполным распадом молочного сахара (лактозы) происходит накопление в крови и тканях веществ (галактозы, галактозо-1-фосфата и галактита), обладающих токсическим действием.
Клиническая картина. Ребенок рождается – абсолютно нормальным. Первые симптомы галактоземии (рвота, падение веса, желтуха) могут появиться вскоре после рождения, как только ребёнок начинает получать молоко – у ребенка происходит интоксикация.
Буквально – через 10-15 дней – у ребенка начинаются боли в желудочно-кишечном тракте; вздутие живота, увеличение печени; на теле – диатез. Потому что ребенок не переносит молоко.
Характерными признаками галактоземии являются: возникает возбуждение; бессонница; судороги; снижение массы тела, жировая дистрофия и цирроз печени, желтуха, катаракта, задержка психомоторного развития. При тяжелой форме галактоземии дети часто погибают ни первом году жизни вследствие нарушений функций печени (цирроз печени) или пониженной сопротивляемости инфекциям.
При тяжёлой форме - смерть наступает через 3-5 месяцев. При лёгкой форме заболевания симптомы менее выражены, но, как правило, отмечаются рвота, задержка роста и развития, катаракта.
Фруктозурия
Это тоже наследственное заболевание. Наследуется аутосомно-рецессивно (т.е. через поколение).
При этом заболевании – в организме: нет фермента, который должен расщеплять фруктовый сахар.
Клиническая картина. Ребенок рождается – абсолютно нормальным, развивается - нормально. Первые симптомы могут появиться как только ребёнок начинает получать фрукты, соки – у ребенка происходит непереносимость всех фруктов.
У ребенка начинаются - вздутие живота, на теле – диатез. Ребенок становится раздражительным – у него возникает – рвота.
Если не прекратить кормить его фруктами – то: возникает задержка развития + судорожный синдром (вплоть до потери сознания.)
ПРОФИЛАКТИКА: исключить из питания фрукты.
III. Болезни накопления:
· Мукополисахаридозы – это нарушения связанные с нарушением соединительной ткани
Синдром Марфана
обусловлен пороком развития соединительной ткани и характеризуется поражением:
а) опорно–двигательного аппарата: высокий рост, диспропорция в росте туловища и конечностей, кисти и стопы длинные с тонкими «паукообразными пальцами», грудная клетка килевидной или воронкообразной формы, кифоз, сколиоз, широкие межреберные промежутки, тонкие и длинные ребра, которые имеют отвесное направление; «птичье» выражение лица (узкий череп, подбородок срезан или выступает, близко посаженные глаза, ушные раковины тонкие и малоэластичные); перерастяженность сухожилий и суставов, слабость связок, мышечная гипотония, недоразвитие подкожной клетчатки;
б) глаз: миопия, голубые склеры, частичный или полный подвывих хрусталика, колобома радужной оболочки;
в) внутренних органов: сердечно–сосудистая система (пороки сердца, крупных сосудов, расслаивающаяся аневризма аорты, аномалия расположения сосудов), уменьшение числа долей легких.
Умственное развитие при этом заболевании обычно не страдает.
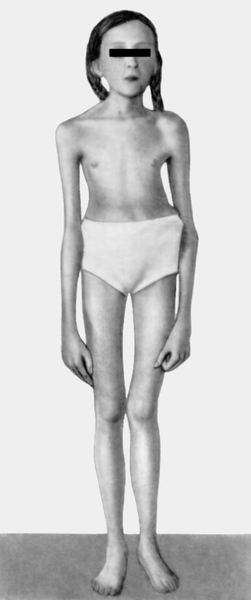 Синдром Марфана встречается с частотой 1: 10000.
Синдром Марфана встречается с частотой 1: 10000.

Болезнь Гунтера (Гурлера)Cиндром Гурлер (гаргоилизм)
К Гаргоилизму относят первые два типа мукополисахаридозов — синдром Гурлер и синдром Гунтера. Джервис (G. A. Jervis, 1950) По данным ВОЗ (1968), синдром Гунтера встречается в 1 случае на 200 000 рождений.