

Типы оградительных сооружений в морском порту: По расположению оградительных сооружений в плане различают волноломы, обе оконечности...

Общие условия выбора системы дренажа: Система дренажа выбирается в зависимости от характера защищаемого...
Типы оградительных сооружений в морском порту: По расположению оградительных сооружений в плане различают волноломы, обе оконечности...
Общие условия выбора системы дренажа: Система дренажа выбирается в зависимости от характера защищаемого...
Топ:
История развития методов оптимизации: теорема Куна-Таккера, метод Лагранжа, роль выпуклости в оптимизации...
Процедура выполнения команд. Рабочий цикл процессора: Функционирование процессора в основном состоит из повторяющихся рабочих циклов, каждый из которых соответствует...
Генеалогическое древо Султанов Османской империи: Османские правители, вначале, будучи еще бейлербеями Анатолии, женились на дочерях византийских императоров...
Интересное:
Берегоукрепление оползневых склонов: На прибрежных склонах основной причиной развития оползневых процессов является подмыв водами рек естественных склонов...
Искусственное повышение поверхности территории: Варианты искусственного повышения поверхности территории необходимо выбирать на основе анализа следующих характеристик защищаемой территории...
Финансовый рынок и его значение в управлении денежными потоками на современном этапе: любому предприятию для расширения производства и увеличения прибыли нужны...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Содержание
Введение
1. Реакции C-ацилирования
1.1 Реакция алкил-де-галогенирования
1.2 Реакция алкил-де-ацилокси-замещения
1.3 Реакции ацилирования кетонов ангидридами
1.4 Реакция ацилирования Фриделя-Крафтса
1.5 Реакции ацилирования сложных эфиров сложными эфирами. Конденсации Кляйзена и Дикмана
2.Реакции N-ацилирования
2.1 Ацилирование аминов ацилгалогенидами
2.2 Ацилирование аминов ангидридами
2.3 Ацилирование аминов карбоновыми кислотами
2.4 Ацилирование аминов сложными эфирами
2.5 Ацилирование аминов амидами
3. Реакции O-ацилирования
3.1 Гидролиз ацилгалогенидов
3.2 Гидролиз ангидридов
3.3 Гидролиз сложных эфиров
3.4 Гидролиз амидов
3.5 Алкоголиз ацилгалогенидов
3.6 Алкоголиз ангидридов
3.7 Этерификация кислот
3.8 Алкоголиз сложных эфиров. Переэтерификация
3.9 Ацилирование карбоновых кислот ацилгалогенидам
3.10 Ацилирование карбоновых кислот кислотами
Заключение
Список литературы
Введение
Ацилирование – введение ацильной группы (ацила) RC в молекулу органического соединения путем замещения атома водорода. В широком смысле ацилирование это замещение любого атома или группы атомов на ацила. В зависимости от атома к которому присоединяют ацил различают C-, N-, O-, S – ацилирование.Реакции ацилирования обладают очень многими полезными свойствами. Они позволяют вести в молекулу функциональную группу C=O путем реакций присоединения либо замещения, не подвергая исходную молекулу окислению (восстановлению). Таким образом, можно получать соединения различных классов: а) амиды; б) сложные эфиры; в) ангидриды карбоновых кислот; г) кетоны и другие полезные соединения. Неудивительно, что реакции ацилирования находят широкое применение в промышленности и в химических исследованиях. В своей курсовой работе я рассмотрю три наиболее важных типа реакций ацилирования C-ацилирование, O-ацилирование и N-ацилирование.
|
|
Реакции C-ацилирования
Наиболее часто в реакциях С-ацилирования используются металлоорганические соединения (реактивы Гриньяра, кислоты Льюиса, соединения алкила с металлом, алкоголяты металлов, а также комплексные соли с алкильными лигандами).
Реакция алкил-дегалогенирования
Рассмотрим реакцию алкил-д-галогенирования (превращения ацилгалогенидов в кетоны с помощью металлоорганических соединений). Ацилгалогениды гладко и в мягких условиях взаимодействуют с диалкилкупратами лития, давая с высокими выходами кетоны. Это происходит по следующей схеме:
 |
Группа R' может быть первичной, вторичной или третичной алкильной или арильной; она может содержать йодо-, кето-, нитро-, циано- и сложноэфирные группы. Успешно проведены реакции, в которых группа R была метильной, первичной алкильной и винильной. Вторичные и третичные алкильные группы можно ввести, если вместо R2CuLi использовать PhS(R) CuLi. Группа R может быть и алкинильной, если в качестве реагента применяется ацетиленид меди R’’CºCСu.
Другой тип металлоорганических реагентов, которые дают хорошие выходы кетонов при обработке ацилгалогенидами, – это кадмийорганические соединения R2Cd (получаемые из реактивов Гриньяра) В этом случае группа R может быть арильной или первичной алкильной. Вторичные и третичные алкилкадмиевые реагенты оказываются, как правило, недостаточно устойчивыми, чтобы служить полезными реагентами в этой реакции. Как в R’COX, так и в R2Cd может присутствовать сложноэфирная группа. Цинкорганические соединения ведут себя аналогично, но используются реже. Ртутьорганические соединения и тетраалкилсиланы также вступают в эту реакцию при катализе AlX3.
Реакции N-ацилирования
|
|
Ацилирование аминов амидами
Это реакция обмена, и ее обычно проводят с солью амина. Уходящей группой служит, как правило, NH2, а не NHR или NR2; в качестве реагентов наиболее широко применяются первичные амины (в виде солей).
Для образования комплекса с уходящим аммиаком можно добавлять BF3. Эту реакцию часто применяют для получения замещенных производных мочевины из самой мочевины:
 |
Диметилформамид можно превратить в другие формамиды продолжительным нагреванием с первичным или вторичным амином.
 |
Реакции O-ацилирования
Гидролиз ацилгалогенидов
Ацилгалогениды очень реакционноспособны, поэтому гидролиз проходит легко. Большинство галогеноангидридов простых кислот следует хранить в безводных условиях, так как они реагируют с влагой воздуха. Поэтому обычно вода оказывается достаточно сильным нуклеофилом для проведения реакции гидролиза, хотя в отдельных случаях необходимо использовать гидроксид-ион.
Реакция, как правило, не имеет синтетической ценности, так как ацилгалогениды обычно получают из кислот. Реакционная способность ацилгалогенидов изменяется в следующем ряду: F<Cl<Br<I. При использовании в качестве нуклеофила карбоновой кислоты возможна реакция обмена.
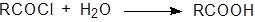 |
Гидролиз галогеноангидридов обычно не катализируется кислотами, за исключением ацилфторидов, когда образование водородной связи может способствовать отщеплению фтора.
Гидролиз ангидридов
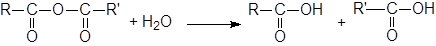 |
Гидролиз ангидридов можно катализировать основаниями. Конечно, OH-группа атакует более энергично, чем вода, но и другие основания могут катализировать эту реакцию. Это явление, называется нуклеофильным катализом.
Гидролиз сложных эфиров
Гидролиз сложных эфиров обычно катализируется как кислотами, так и основаниями. Поскольку группа OR обладает более слабыми нуклеофугными свойствами, чем галогены или OCOR, вода не гидролизует большинство сложных эфиров.
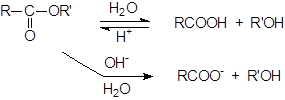 |
При катализе основаниями, атакующей частицей служит более сильный нуклеофил – OH-группа. Эта реакция носит название омыления и приводит к соли кислоты. Кислоты катализируют реакцию за счет того, что положительный заряд атома углерода карбонильной группы становится больше, и, следовательно, он легче подвергается атаке нуклеофилом. Обе реакции обратимы, и поэтому практической ценностью обладают только тогда, когда равновесия удаётся каким либо образом сместить вправо. А поскольку образование соли – один из таких способов, гидролиз сложных эфиров в препаративных целях почти всегда проводят в щелочных растворах, за исключением тех случаев, когда вещество неустойчиво к действию оснований.
|
|
Гидролиз амидов
Незамещенные амиды RCONH2 способны гидролизоваться под действием как кислотных, так и основных катализаторов; при этом образуются соответствующая свободная кислота и ион аммония или соль кислоты и аммиак.
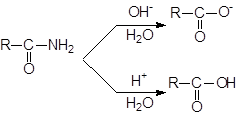 |
Аналогично можно гидролизовать N-замещенные (RCONHR’) и N, N-дизамещенные (RCONR’2) амиды, причем вместо аммиака получаются первичные и вторичные амины соответственно (или их соли). Вода является слишком слабым нуклеофилом для гидролиза большинства амидов, так как группа NH2 обладает еще более низкими нуклеофугными свойствами, чем группа OR. Даже в условиях кислотного или основного катализа часто требуется длительное нагревание.
Алкоголиз ацилгалогенидов
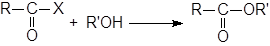 |
Для связывания образующегося НХ часто добавляют основания. В методе Шоттена-Баумана используется водный раствор щелочи, но часто применяется пиридин. Как R, так и R’ могут быть первичными, вторичными или третичными алкилами или арилами. В сложных случаях, особенно для стерически затрудненных кислот или третичных R’, вместо спирта можно брать алкоголят ион.
При использовании в качестве ацилгалогенидов фосгена можно получить галоформные эфиры или карбонаты:
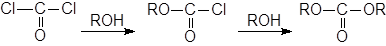 |
Важный пример – это синтез из фосгена и бензилового спирта карбобензоксихлорида (PhCH2OCOCl), который широко применяется для защиты аминогрупп в пептидном синтезе.
|
|
Алкоголиз ангидридов
Диапазон применяемости этого метода такой же, как и реакции алкоголиза ацилгалогенидов. И хотя ангидриды немного менее реакционно способны, чем ацилгалогениды, их часто используют для получения сложных эфиров.
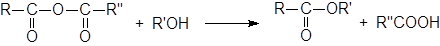 |
В качестве катализаторов применяют кислоты, кислоты Льюиса и основания, но наиболее часто – пиридин. Реакция циклических ангидридов приводит к моноэтерифицированным дикарбоновым кислотам, например:
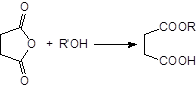 |
Этерификация кислот спиртами представляет собой реакцию, обратную реакции гидролиза сложных эфиров:
 |
Ее можно осуществить только тогда, когда равновесие удается сместить вправо. Для этой цели имеется много способов, среди которых:
– прибавление одного из реагентов (обычно спирта) в избытке;
– удаление эфира или воды отгонкой;
– азеотропная отгонка воды;
Удаление воды, используя водоотнимающие средства или молекулярные сита.
Если R’ = метил, то наиболее общий способ смещения равновесия – это добавление избытка МеОН, а если R’ = этил, то предпочтительнее удалять воду азеотропной отгонкой. В качестве катализаторов чаще всего используются серная кислота и TsOН, хотя в случае некоторых активных кислот (например муравьиной или трифтороуксусной) катализатора не требуется. Группа R’ может быть не только метильной или этильной, но также и другой первичной или вторичной алкильной группой, однако третичные спирты обычно образуют карбокатионы и происходит элиминирование. для получения эфиров фенолов можно использовать сами фенолы, но выходы, как правило очень низкие. Этерификация катализируется кислотами (но не основаниями).
Другим способом получения сложных эфиров из кислоты является обработка спиртом в присутствии дегидратирующих веществ, одним из которых является дициклогексилкарбодиимид, в ходе реакции превращающийся в дициклогексилмочевину:
 |
3.8 Алкоголиз сложных эфиров. Переэтерификация
Переэтерификация катализируется кислотами или основаниями. Это обратимая реакция и равновесие необходимо смещать в желаемую сторону.
Во многих случаях низкокипящие эфиры можно превратить в более высококипящие путем отгонки низкокипящего спирта по мере его образования. Эта реакция была использована как метод ацилирования первичных ОН-групп в присутствии вторичных ОН – групп.
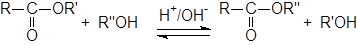 |
Заключение
Итак, после подробного изучения реакций ацилирования: C-ацилирование, O-ацилирование, N-ацилирование, а именно с получением сложных эфиров, кетонов и амидов можно сделать вывод, что эти реакции действительно играют важную роль в химических исследованиях и промышленности, а также обладают очень многими полезными свойствами. Именно поэтому реакции ацилирования требуют очень серьезного контроля, точности в применении химических веществ. Естественно существуют реакции, которые идентичны по своему конечному результату, но одни из них наиболее удобны в применении, другие наименее, поэтому и необходимо их тщательное изучение.
|
|
Список литературы
1. Органическая химия Джерри Марч; Москва «Мир» 1987;
2. Механизмы реакций в органической химии» Питер Сайкс; Химия 1971;
3. Органическая химия Ю.С. Шабаров; «Химия» 1994;
4. Основы органической химии Франк Л. Вайзман; «Химия» 1995.
5. Химическая энциклопедия т. 1 с. 442 издательство «Советская энциклопедия». 1988 – 623 с.: ил.6. А. Терней «Современная органическая химия» т. 2 с. 221 издательство «Мир», 1981 – 655 с.7. «Органикум» т. 1 издательство «Мир» 1992 – 488 с.8. «Органикум» т. 2 издательство «Мир» 1992 – 472 с.9. А.А. Петров, Х.В. Бальян, А.Т. Трощенко; «Органическая химия», учебник для вузов.
10. Марч И. «Органическая химия».
11. Моррисон Р. Бойд Р. «Органическая химия».
12. Робертс Дж., Касерио М. «Основы органической химии».
Содержание
Введение
1. Реакции C-ацилирования
1.1 Реакция алкил-де-галогенирования
1.2 Реакция алкил-де-ацилокси-замещения
1.3 Реакции ацилирования кетонов ангидридами
1.4 Реакция ацилирования Фриделя-Крафтса
1.5 Реакции ацилирования сложных эфиров сложными эфирами. Конденсации Кляйзена и Дикмана
2.Реакции N-ацилирования
2.1 Ацилирование аминов ацилгалогенидами
2.2 Ацилирование аминов ангидридами
2.3 Ацилирование аминов карбоновыми кислотами
2.4 Ацилирование аминов сложными эфирами
2.5 Ацилирование аминов амидами
3. Реакции O-ацилирования
3.1 Гидролиз ацилгалогенидов
3.2 Гидролиз ангидридов
3.3 Гидролиз сложных эфиров
3.4 Гидролиз амидов
3.5 Алкоголиз ацилгалогенидов
3.6 Алкоголиз ангидридов
3.7 Этерификация кислот
3.8 Алкоголиз сложных эфиров. Переэтерификация
3.9 Ацилирование карбоновых кислот ацилгалогенидам
3.10 Ацилирование карбоновых кислот кислотами
Заключение
Список литературы
Введение
Ацилирование – введение ацильной группы (ацила) RC в молекулу органического соединения путем замещения атома водорода. В широком смысле ацилирование это замещение любого атома или группы атомов на ацила. В зависимости от атома к которому присоединяют ацил различают C-, N-, O-, S – ацилирование.Реакции ацилирования обладают очень многими полезными свойствами. Они позволяют вести в молекулу функциональную группу C=O путем реакций присоединения либо замещения, не подвергая исходную молекулу окислению (восстановлению). Таким образом, можно получать соединения различных классов: а) амиды; б) сложные эфиры; в) ангидриды карбоновых кислот; г) кетоны и другие полезные соединения. Неудивительно, что реакции ацилирования находят широкое применение в промышленности и в химических исследованиях. В своей курсовой работе я рассмотрю три наиболее важных типа реакций ацилирования C-ацилирование, O-ацилирование и N-ацилирование.
Реакции C-ацилирования
Наиболее часто в реакциях С-ацилирования используются металлоорганические соединения (реактивы Гриньяра, кислоты Льюиса, соединения алкила с металлом, алкоголяты металлов, а также комплексные соли с алкильными лигандами).
Реакция алкил-дегалогенирования
Рассмотрим реакцию алкил-д-галогенирования (превращения ацилгалогенидов в кетоны с помощью металлоорганических соединений). Ацилгалогениды гладко и в мягких условиях взаимодействуют с диалкилкупратами лития, давая с высокими выходами кетоны. Это происходит по следующей схеме:
|
Группа R' может быть первичной, вторичной или третичной алкильной или арильной; она может содержать йодо-, кето-, нитро-, циано- и сложноэфирные группы. Успешно проведены реакции, в которых группа R была метильной, первичной алкильной и винильной. Вторичные и третичные алкильные группы можно ввести, если вместо R2CuLi использовать PhS(R) CuLi. Группа R может быть и алкинильной, если в качестве реагента применяется ацетиленид меди R’’CºCСu.
Другой тип металлоорганических реагентов, которые дают хорошие выходы кетонов при обработке ацилгалогенидами, – это кадмийорганические соединения R2Cd (получаемые из реактивов Гриньяра) В этом случае группа R может быть арильной или первичной алкильной. Вторичные и третичные алкилкадмиевые реагенты оказываются, как правило, недостаточно устойчивыми, чтобы служить полезными реагентами в этой реакции. Как в R’COX, так и в R2Cd может присутствовать сложноэфирная группа. Цинкорганические соединения ведут себя аналогично, но используются реже. Ртутьорганические соединения и тетраалкилсиланы также вступают в эту реакцию при катализе AlX3.
|
|
|
История развития пистолетов-пулеметов: Предпосылкой для возникновения пистолетов-пулеметов послужила давняя тенденция тяготения винтовок...

Семя – орган полового размножения и расселения растений: наружи у семян имеется плотный покров – кожура...

История развития хранилищ для нефти: Первые склады нефти появились в XVII веке. Они представляли собой землянные ямы-амбара глубиной 4…5 м...

Состав сооружений: решетки и песколовки: Решетки – это первое устройство в схеме очистных сооружений. Они представляют...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!