Повреждение клеток характеризуется развитием разнообразных изменений в них. Однако их можно объеденить в несколько групп:
ь Дистрофии.
ь Дисплазии.
ь Типовые нарушения субклеточных структур и компонентов.
ь Некроз.
Дистрофии (от лат. dys - нарушение, расстройство + греч. trophe - питаю) - это нарушения обмена веществ в клетках, сопрвождающиеся расстройствами их функций, пластических процессов и структурными изменениями, ведущими к нарушению их жизнедеятельности.
Основными механизмами дистрофий являются:
· синтез аномальных веществ в клетке, например, белково-полисахаридного комплекса амилоида;
· избыточная трансформация одних соединений в другие, например, жиров и углеводов в белки, углеводов в жиры;
· декомпозиция (фанероз), например, белково-липидных комплексов мембран;
· инфильтрация клеток (и межклеточного вещества) органическими и неорганическими соединениями, например, холестерином и его эфирами стенок артерий при атеросклерозе.
К числу основных разновидностей клеточных дистрофий в зависимости от преимущественно нарушенного вида обмена веществ относят:
ь белковые (диспротеинозы);
ь жировые (липидозы);
ь углеводные;
ь пигментные;
ь минеральные.
Диспротеинозы. Характеризуются изменением фихико-химических свойств белков клеток и как следствие нарушением их ферментативной и структурной функций. Наиболее часто диспротеинозы проявляются в виде зернистой, гиалиново-капельной и гидропической дистрофии. Нередко они представляют собой последовательные этапы нарушения обмена цитоплазматических белков, приводящих к некрозу клеток.
При зернистой дистрофии в цитоплазме появляются гранулы (зерна) белка. (см. рис. 6.3.). Они образуются в результате инфильтрации (проникновения) его из межклеточной жидкости, трансформации углеводов и жиров в белки, распада (декомпозиции) липопротеидов цитоплазмы и мембран. Одной из главных общих причин зернистой дистрофии является нарушение энергообеспечения клеток.
Гиалиновая дистрофия характеризуется накоплением в цитоплазме белковых гиалиноподобных ацидофильных включений ("капель"). Одновременно с этим выявляются признаки деструкции клеточных органелл. Признаки гиалиновой дистрофии наблюдаются при состояниях, вызывающих повышение проницаемости клеточных мембран.
Гидропическая (водяночная, вакуольная) дистрофия является результатом такого изменения физико-химических свойств белков цитоплазмы, которое сопровождается повышением онкотического давления в клетке и избыточной гидратацией белковых мицелл. В цитоплазме клеток формируются вакуоли, наполненные жидкостью и не содержащие липидов или гликогена. При электронной микроскопии обнаруживаются признаки внутриклеточного отека и набухания органелл. Наиболее частыми причинами гидропической дистрофии являются гипоксия, воздействие ионизирующей радиации, токсины микроорганизмов и паразитов, нарушения питания. 
Рис. 6.3. Диспротеинозы.
Липидозы. К липидозам относят различные по химическому составу вещества, нерастворимые в воде. Липидозы проявляются либо увеличением содержания внутриклеточных липидов, либо появлением их в клетках, где они в норме отсутствуют, либо образованием липидов аномального химического состава. Липидозы, так же, как и диспротеинозы, наиболее часто наболюдаются в клетках сердца, печени, почек, мозга и носят соответсвующие названия (жировая дистрофия сердца, печени, почек, мозга).
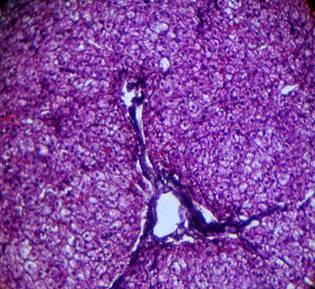

Рис. 6.4. Липидоз печени
Углеводные дистрофии. Характеризуются нарушением обмена полисахаридов (гликогена, мукополисахаридов) и гликопротеидов (муцина, мукоидов).
"Полисахаридные" дистрофии проявляются:
1) уменьшением их содержания в клетке (например, гликогена при сахарном диабете);
2) их отсутствием или значительным снижением (агликогенозы);
3) накоплением их избытка (гликогенная инфильтрация клеток, гликогенозы).
Причиной углеводных дистрофий чаще всего являются эндокринопатии (например, инсулиновая недостаточность) или ферментопатии (отсутствие или низкая активность ферментов, принимающих участие в процессах синтеза и распада углеводов).
Углеводные дистрофии, связанные с нарушением метаболизма гликопротеидов, характеризуются, как правило, накоплением муцинов и мукоидов, имеющих слизистую консистенцию. В связи с этим их называют слизистыми дистрофиями. (см. рис. 6.4.) Причинами их наиболее часто служат эндокринные расстройства (например, недостаточная продукция или низкая активность гормонов щитовидной железы), а также прямое повреждающее действие на клетки патогенных факторов.
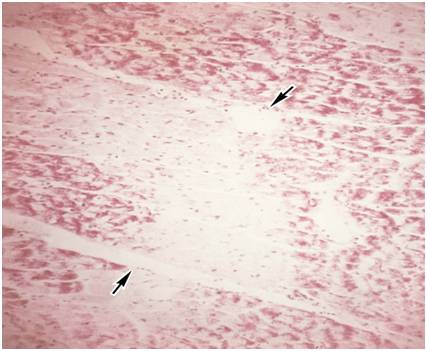
Рис. 6.5. Микропрепарат сердца при углеводной дистрофии миокарда: стрелками указан крупный очаг сниженного содержания гликогена в кардиомиоцитах; по периферии очага видны кардиомиоциты с гранулами гликогена красного цвета.
Пигментные дистрофии (диспигментозы). Пигменты клеток организма человека и животных принимают участие в реализации многих функций: синтез и катаболизм веществ, рецепция различных воздействий, защита от повреждающих факторов.
Клеточные пигменты являются хромопротеидами, т.е. соединениями, состоящими из белка и красящего вещества (см. рис. 6.6)
В зависимости от биохимического строения эндогенные клеточные пигменты разделяют следующим образом:
- гемоглобиногенные (ферритин, гемосидерин, билирубин, гематоидин, гематин, порфирин);
- протеиногенные, тирозиногенные (меланин, адренохром, пигменты охроноза и энтерохромафинных клеток);
- липидогенные, липопротеиногенные (липофусцин, гемофусцин, цероид, липохромы).

Рис. 6.6. Депигментоз сетчатки
Все диспигментозы делятся на несколько групп в зависимости от их происхождения, механизма развития, биохимической структуры пигмента, проявлений и распространенности.
Виды диспигментозов
По происхождению:
1. Первичные (наследственные, врожденные).
2. Вторичные, приобретенные (возникающие под действием патогенных агентов в течение постнатального периода жизни организма).
По механизму развития:
1. Обусловленные дефектами ферментов (ферментопатиями) метаболизма пигмента и (или) изменением их активности.
2. Связанные с изменением содержания и (или) активности ферментов транспорта пигментов через мембраны клетки.
3. Вызванные повреждением мембран клеток.
4. Обусловленные накоплением избытка пигментов в клетках, обладающих свойством фагоцитоза.
По биохимической структуре пигмента:
1. Гемоглобиногенные, "железозависимые".
2. Протеиногенные, тирозиногенные.
3. Липидогенные, липопротеиногенные.
По проявлениям:
1. Появление в клетке пигмента, отсутствующего в ней в норме.
2. Накопление избытка пигмента, образующегося в клетке в норме.
3. Уменьшение количества пигмента, образующегося в клетке в норме.
По распространенности:
1. Местные (регионарные).
2. Общие (распространенные).
Гемоглобиногенные диспигментозы включают гемосидероз, гемохроматоз, гемомеланоз, порфирию, накопление избытка прямого билирубина в гепатоцитах.
Большинство гемоглобиногенных пигментов относятся к продуктам катаболизма гемоглобина. Некоторые из них (ферритин, гемосидерин) образуются с участием железа, всасывающегося в кишечнике.
Часть гемоглобиногенных диспигментозов является результатом ферментопатий. К ним относятся, в частности, первичный гемохроматоз и порфирия.
Первичный гемохроматоз - заболевание, обусловленное генетическим дефектом (передается аутосомно-доминантным путем) группы ферментов, участвующих в процессах транспорта железа из полости кишечника. При этом в кровь поступает избыток железа, которое накапливается в виде ферритина и гемосидерина в клетках различных тканей и органов (печени, миокарда, кожи, желез внутренней секреции, слюнных желез и др.). Сходные изменения наблюдаются и при вторичном гемохроматозе. Он является результатом либо приобретенной недостаточности ферментов, обеспечивающих обмен пищевого железа (при алкоголизме, интоксикациях), либо - повышенного поступления железа в организм с продктами питания или железосодержащими лекарственными препаратами, либо следствием избыточного гемолиза эритроцитов.
Порфирия характеризуется накоплением в клетках уропорфириногена I, порфобилина, порфириногенов. Одной из частых причин порфипии является дефицит или низкая кинетическая активность ферментов метаболизма порфиринов (в частности, уропорфириноген-III-косинтетазы) наследственного или приобретенного характера.
Большинство других разновидностей гемоглобиногенных диспигментозов (гемосидероз, гемомеланоз) являются следствием избыточного накопления пигметов в клетках в связи с повышенным гемолизом эритроцитов различного генеза (при инфекциях, интоксикациях, переливании иногруппной крови, резус-конфликте и др.).
Протеиногенные (тирозиногенные) диспигментозы проявляются усилением или ослаблением пигментации тканей (локального или общего характера) продуктами метаболизма тирозина.
Усиление пигментации нередко является следствием избытка в клетках меланина (меланоз, от греч. melas - темный, черный). Наблюдается при надпочечниковой недостаточности, обусловленной уменьшением их массы, например, при туберкулезном или опухолевом поражении; при аденоме гипофиза, гипертиреоидизме, опухолях яичников. Считают, что избыток меланина в клетках является результатом его повышенного синтеза из тирозина вместо адреналина. Процесс меланинообразования потенциируется АКТГ, уровень которого повышен в условиях дефицита адреналина в крови.
Накопление пигмента охроноза (от греч. ochros - желтый, желтоватый) в клетках наблюдается при первичной (наследственной) ферментопатии, характеризующейся недостаточностью энзимов метаболизма тирозина и фенилаланина. При этом гиперпигментация носит местный или распространенный характер. Пигмент накапливается в клетках тканей носа, ушных раковин, склер, трахеи, бронхов, сухожилий, хрящей и др.
Ослабление пигментации тканей или отсутствие пигмента в их клетках (альбинизм, от лат. albus - белый) также может быть первичного или вторичного происхождения. При альбинизме меланин отсутствует в клетках кожи, радужки глаз, в волосах. Причиной этого чаще всего является наследственно обусловленное отсутствие в клетках фермента тирозииназы. В случае местного уменьшения пигментации, например, кожи (лейкодерма, витилиго) существенное значение имеет вторичное нарушение обмена меланина в связи с нейроэндокринными нарушениями его регуляции (при гипоинсулинизме, снижении уровня гормонов паращитовидных желез), вследствие образования антител к меланину либо в результате повышенного разрушения меланоцитов при воспалении или некрозе тканей.
Липидогенные диспигментозы, характеризующиеся чаще всего увеличением в клетках количества пигментов липидного или липопротеидного характера (липофусцина, гемофусцина, липохромов, цероида). Все эти пигменты весьма сходны по основным физическим и биохимическим свойствам. У человека обычно встречаются различные варианты местного липофусциноза наследственного (реже) или приобретенного (чаще) происхождения.
Считается, что основными причинами приобретенного липофусциноза являются гипоксия тканей, дефицит в организме витаминов, белка, отдельных видов липидов. Наиболее часто он развивается в пожилом и старческом возрасте, у людей с хроническими "обменными" заболеваниями.
Наследственные и врожденные липофусцинозы характеризуются накоплением избытка липофусцина в клетках, сочетающимся обычно с ферментопатиями (т.е. эти липофусцинозы являются вариантом болезней накопления - тезаурисмозов). Примерами этих болезней могут быть нейрональные липофусцинозы (отложение избытка липофусцина в нейронах, что сочетается со снижением интеллекта, зрения, слуха, развитием судорог); печеночные липофусцинозы, сочетающиеся с нарушениями обмена билирубина, обусловленными наследственными дефектами ферментов транспорта глюкоронизации желчных пигментов.
Минеральные дистрофии. Проявляются значительным уменьшением или увеличением сордержания минеральных веществ в клетках. Наибольшее значение имеют нарушения обмена соединений кальция, калия, железа, цинка, меди. Их ионизированные и молекулярные фракции участвуют в процессах регуляции проницаемости мембран клеток, активности ферментов, формирования потенциала покоя и действия, реализации действия гормонов и нейромедиаторов, электромеханического сопряжения в в миоцитах и многих других.
Минеральные дистрофии характеризуются накоплением избыточного содержания в клетках молекулярных или ионизированных фракций катионов (например, кальцинозы, сидерозы, отложения меди при гепатоцеребральной дистрофии) или уменьшением их содержания.
Одной из наиболее распространенных у человека разновидностей клеточных минеральных дистрофий является кальциноз - накопление ("отложение") избытка солей кальция в клетках. Кальциноз может носить общий или местный характер. На "территории" клетки в наибольшей мере соли кальция накапливаются в митохондриях, лизосомах (фаголизосомах), в канальцах саркоплазматической сети. Основной причиной клеточного кальциноза является изменение физико-химических свойств гиалоплазмы клетки (например, внутриклеточный алкалоз), сочетающееся с абсорбцией кальция. Наиболее часто отмечается кальциноз клеток миокарда, эпителия почечных канальцев, легких, слизистой желудка, стенок артерий.
К числу дистрофий относят также тезаурисмозы (от греч. thesauriso-накопление, поглощение, наполнение). Они характеризуются накоплением избытка различных веществ в клетках, что сопровождается нарушением их структуры и функции, а также - интенсивности и характера метаболических и пластических процессов в них.
Практически все тезаурисмозы - результат наследственной патологии ферментов, передающихся, как правило, по аутосомно-рецессивному типу. Наследуемые изменения в генетической программе обусловливают дефект ферментов (лизосомальных, мембраносвязанных, свободных). Следствием этого является нарушение метаболизма в клетке, обусловливающее накопление в ней продуктов неполного или аномального расщепления субстратов.
В зависимости от биохимической структуры накапливающихся в клетках веществ тезаурисмозы разделяют на липидные (липидозы), гликогеновые (гликогенозы) аминокислотные, нуклеопротеидные, мукополисахаридные, муколипидные. Наиболее распространенными разновидностями тезаурисмозов являются липидные и гликогеновые.
Дисплазии (от dys - нарушение, расстройство + греч. plasis - образую) - общее название нарушений процесса развития (дифференцировки, специализации) клеток, проявляющихся стойким изменением их структуры и функции, что ведет к расстройству их жизнедеятельности.
Причинами дисплазий являются факторы физического, химического или биологического характера, повреждающие геном клетки. При этом нарушается генетическая программа клеток или механизмы ее реализации. Именно это обусловливает стойкие и, как правило, наследуемые от клетки к клетке изменения в отличие от дистрофий, которые нередко носят временный, обратимый характер и могут устраняться при прекращении действия причинного фактора.
Основным механизмом дисплазий является расстройство процесса дифференцировки, который заключается в формировании структурной и функциональной специализации клетки. Клеточная дифференцировка определяется в основном генетической программой. Однако реализация этой программы в существенной мере зависит от сложных взаимодействий ядра и цитоплазмы, микроокружения клетки, влияния на нее БАВ и многих других факторов. Именно поэтому даже при одном и том же изменении в геноме различных клеток проявления дисплазий могут носить "разноликий характер".
Дисплазии проявляются изменением величины и формы клеток, их ядер и других органелл, числа и строения хромосом. Как правило, клетки увеличены в размерах, имеют неправильную, причудливую форму ("клетки-монстры"), соотношение различных органелл в них диспропорционально. Нередко в таких клетках обнаруживаются различные включения, признаки дистрофических процессов.
В качестве примеров клеточных дисплазий можно назвать образование мегалобластов в костном мозге при пернициозной анемии, серповидных эритроцитов при наличии патологического гемоглобина, крупных нейронов - "монстров" при поражении коры большого мозга (туберкулезный склероз), многоядерных гигантских клеток с причудливым расположением хроматина при нейрофиброматозе (болезнь Реклинхаузена). Клеточные дисплазии являются одним из проявлений атипизма опухолевых клеток.
Типовые нарушения субклеточных структур и компонентов.
Клетка представляет собой многокомпонентную систему. Она включает в себя ядро; гиалоплазму; органеллы (митохондрии, пероксиомы, рибосомы, эндоплазматическую сеть, лизосомы, пластинчатый комплекс, или комплекс Гольджи, клеточный центр, микротрубочки, микрофиламенты); ьметаплазматические специализированные специализированные образования (миофибриллы, нейрофибриллы, тонофибриллы, микроворсинки, десмосомы и др.); включения (трофические, секреторные, а также специфические для отдельных клеток, например, гранулы тучных клеток, или лаброцитов, содержащие серотонин, гистамин, гепарин и другие вещества). Указанные компоненты клетки окружены плазмолеммой (цитолеммой) (см. рис. 6.7).
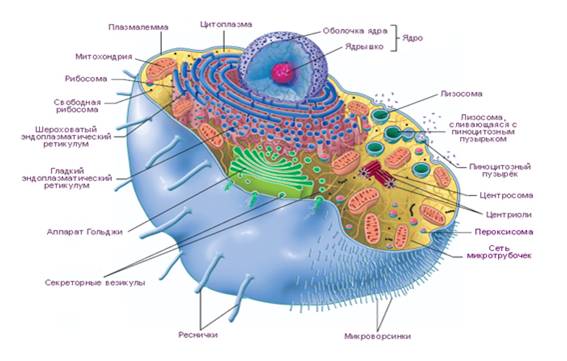
Рис. 6.7. Строение клетки
Повреждение клетки характеризуется большим или меньшим нарушением структуры функции всех ее компонентов. Однако при действии различных патогенных факторов могут преобладать признаки повреждения отдельных из них.
Ядро является "носителем" генетической программы клетки. Повреждение ядра сочетается с изменением его величины и формы, числа ядрышек в нем, конденсацией хроматина по периферии ядра (маргинация хроматина), нарушением двухконтурности или разрывами ядерной оболочки, слиянием ее с полоской маргинации хроматина, появлением включений, спутников ядра и др.
Митохондрии. Эти органеллы участвуют во многих внутриклеточных процессах. Главными из них являются окисление, сопряженное с фосфорилированием, ведущее к образованию АТФ и регуляции внутриклеточного содержания кальция (митохондрии обладают высокой кальциевой емкостью), калия, ионов водорода.
При действии патогенных факторов отмечается изменение общего числа митохондрий, а также их структуры и отдельных органелл. Уменьшение числа митохондрий по отношению к общей массе клетки, в частности в печени, наблюдается при длительном голодании, после облучения организма, при сахарном диабете.
Стереотипными для действия большинства повреждающих факторов изменениями отдельных митохондрий являются уменьшение или увеличение их размеров и изменение формы. Многие патогенные воздействия на клетку (гипоксия, эндо- и экзогенные токсические агенты, в том числе лекарственные препараты при их передозировке, ионизирующая радиация, изменение осмотического давления) сопровождаются набуханием и вакуолизацией митохондрий, что может привести к разрыву их мембран, фрагментации и гомогенизации крист. Нередко отмечаются утрата гранулярной структуры и гомогенизация крист. Нередко отмечаются утрата гранулярной структуры и гомогенизация матрикса органелл, потеря двухконтурности их наружной мембраны, отложения в матриксе органических (миелин, липиды, гликоген) и неорганических (чаще всего соли кальция) соединений. Нарушение структуры митохондрий приводит 3к существенному подавлению процесса дыхания в них и образования АТФ, а также к дисбалансу ионов (Са2+, К+, Н+) внутри клетки.
Лизосомы. В норме ферменты лизосом обеспечивают обновление структур клетки при их старении или повреждении, а также уничтожение чужеродных агентов в процессе фагацитоза.
При патогенных воздействиях высвобождение и активация ферментов лизосом может привести к "самоперевариванию" (аутолизу) клетки. Повышенный выход лизосомальных гидролаз в цитоплазму может быть обусловлен механическим разрывом их мембраны или значительным повышением проницаемости ("лабилизацией") последних. Это является следствием накопления в клетках ионов водорода (внутриклеточный ацидоз), воздействия продуктов СПОЛ, токсинов и других агентов.
У человека и животных нередко выявляются также первичные, наследственные нарушения функций лизосом (так называемые лизосомные болезни). Они характеризуются дефицитом и (или) снижением активности лизосомальных ферментов. Это, как правило, сопровождается накоплением в клетке избытка веществ, которые в норме метаболизируются с участием энзимов лизосом. Указанные формы лизосомальных ферментопатий являются разновидностью тезаурисмозов - болезней накопления, к которым относятся как уже указывалось, гликогенозы, ганглиозидозы, некоторые гепатозы (сопровождающиеся накоплением в гепатоццитах липофусцина и, как правило, прямого билирубина) и др.
Рибосомы. Эти органеллы необходимы для реализации генетической программы клеток. С их участием происходит синтез белка на основе считывания информации с и-РНК. Поэтому около 40% массы рибосом составляет РНК. При действии повреждающих факторов наблюдается разрушение группировок субъединиц рибосом (полисом), состоящих обычно из нескольких рибосом - "мономеров"; уменьшение числа рибосом, отрыв органелл от внутриклеточных мембран. Эти изменения сопрвождаются снижением интенсивности синтеза белка в клетке.
Эндоплазматическая сеть. Выполняет в клетке функции накопления и распределения различных веществ (в частности, ионов кальция в миоцитах), а также участвуют в инактивации химических агентов. При повреждении отмечается расширение канальцев сети, вплоть до образования крупных вакуолей и цистерн вследствие накопления в них жидкости; очаговая деструкция мембран канальцев сети, их фрагментация. Изменение структуры эндоплазматической сети может сопровождаться развитием клеточных дистрофий, нарушением распространения импульса возбуждения, сократительной функции мышечных клеток, процессов обезвреживания цитотоксических факторов (ядов, метаболитов, свободных радикалов и др.).
Пероксисомы (микротельца). Топографически тесно связаны с эндоплазматической сетью. В микротельцах содержатся различные оксидазы, участвующие в процессах окисления высших жирных кислот, углеводов, аминокислот и других (в том числе цитотоксических) субстратов расщепления перекиси водорода, различных восстановительных компонентов дыхательной цепи. При повреждениях клетки различного генеза может наблюдаться увеличение (в условиях алкогольной интоксикации, вирусной агрессии) или уменьшение (при гипоксии, действии ионизирующей радиации) числа пероксисом. Известны также первичные нарушения функций пероксисом наследственного происхождения ("пероксисомные болезни"). Они характеризуются нарушением обмена веществ в результате либо дефицита и (или) дефекта отдельных ферментов пероксисом, чаще всего каталазы, либо отсутствия микротелец в клетке.
Комплекс Гольджи. Играет существенную роль в процессах транспорта веществ в клетках с высокой метаболической и секреторной активностью, особенно в железах внутренней секреции и клетках, продуцирующих слизь. В этом комплексе также синтезируется ряд веществ (полисахариды, белки), активируются ферменты, депонируются различные соединения. С его участием "генерируются" лизосомы. Повреждение комплекса Гольджи сопровождается структурными изменениями, сходными с таковыми в эндоплазматической сети. При этом нарушаются выведение из клетки продуктов жизнедеятельности, инактивация в ней токсичных соединений, что может обусловить расстройство ее функции в целом.
Микротрубочки, микрофиламенты, промежуточные филаменты (цитокератины, нейрофиламенты, глиальные нити). Они составляют "скелет" клетки, обеспечивают выполнение ее опорной, транспортной, контрактильной, двигательной функций. Повреждение цитоскелета может обусловить нарушение тока секреторных гранул или жидкостей, реализации фагоцитоза, митотического деления клеток, упорядоченного движения ресничек (например, эпителия дыхательных путей или "хвоста" сперматозоида, являющегося эквивалентом реснички).
Гиалоплазма (цитоплазматический матрикс). Представляет собой жидкую слабовязкую внутреннюю среду клетки. Основными компонентами гиалоплазмы являются внутриклеточная жидкость, различные структуры: органеллы, метаплазматические образования и включения.
Действие на клетку повреждающих факторов может обусловливать уменьшение или увеличение содержания в гиалоплазме жидкости, протеолиз или коагуляцию белка, образование "включений", не встречающихся в норме.
Изменение состояния гиалоплазмы в свою очередь существенно влияет на процессы метаболизма, протекающие в ней, в связи с тем, что многие ферменты (например, гликолиза) находятся в клеточном матриксе; на функцию органелл; на процессы восприятия регулируюших и других влияний на клетку.
Прижизненное изучение клеток показало, что в гиалоплазме наблюдаются упорядоченная циркуляция внутриклеточной жидкости, а также ритмические движения органелл. Высказываются допущения, что в различных регионах клетки и ее органеллах может циркулировать разная по составу жидкость. При повреждениях клеток возможно нарушение упорядоченного характера циркуляции цитоплазматической жидкости. Примером дисциркуляторных расстройств могут быть изменения скорости транспорта нейромедиаторов по аксонам нейронов, замедление миграции фагоцитов (вследствие медленного перемещения гиалоплазмы в псевдоподии), развитие так называемого "парциального" отека в клетках (например, отек ядра, митохондрий, миофибрилл и т.д.).
Плазмолемма. В норме выполняет защитную, барьерную, контактную, информационную, транспортную функции. При повреждении клетки указанные функции плазмолеммы страдают в большей или меньшей мере. Это обусловлено значительными изменениями ее проницаемости (чаще повышением), целостности, числа и чувствительности рецепторных структур, трансмембранных "каналов" и другими отклонениями.
Повреждение отдельной клетки (включая и отдельные ее компоненты) может нарушить межклеточные взаимодействия ("общение") и "кооперацию". В основе этого лежит изменение свойств и (или) структуры плазмолеммы, а также находящихся в ней и на ней рецепторных образований, поверхностных антигенов, межклеточных стыков; отклонение от нормы "набора" и свойств метаболитов, в том числе биологически активных (медиаторов и модуляторов "общения"). Это может потенцировать степень и масштаб расстройств в уже поврежденной клетке, а также обусловить альтерацию других, интактных клеток.
Таким образом, совокупность изменений субклеточных структур и их функций, клеток в целом, а также нарушение их взаимодействия и кооперации лежат в основе развития типовых патологических процессов, типовых форм патологии органов и физиологических систем, конкретных болезней и болезненных состояний.