Кулонометрический анализ основан на определении количества электричества, затраченного для электрохимического превращения исследуемого вещества или электроактивных реагента, продукта химической реакции с участием определяемого компонента.
При введении в раствор двух электродов и электрическом внешнем соединении их возникают электрохимические системы следующих типов:
– электролитическая ванна, потребляющая электрическую энергию из внешней цепи;
– гальванический элемент, отдающий электроэнергию во внешнюю цепь.
В местах контакта проводников I и II рода механизм передачи тока меняется, ионы электролита разряжаются (полностью или частично) на одном из электродов, а нейтральные атомы (группы атомов) приобретают заряд на другом. Химические превращения, сопровождающие эти процессы, называются электролизом. При электролизе более положительный электрод считают анодом, а более отрицательный – катодом. Соответственно названия ионов, движущихся к аноду – анионы, а к катоду – катионы.
Соотношения между количеством прошедшего электричества и массой вещества, выделившегося при электролизе, открыты Майклом Фарадеем (1791-1867) в 1833 г. (I, II законы Фарадея). Они могут быть выражены электрохимическим эквивалентом (1):
 , (1)
, (1)
где М(А) – вес одного моля вещества (или атомная масса грамматома его);
F –постоянная Фарадея, т.е. количество электричества, необходимого для электрохимического превращения 1 г-экв вещества, определяется по произведению (2):
 (2)
(2)
где NA – постоянная Авогадро, моль–1;
ē –элементарный заряд электрона, Кл.
Таким образом, величина F численно равна заряду 1 моля электронов, участвующих в процессе.
При прохождении тока через электрохимическую систему требуется движущая сила или потенциал для преодоления сопротивлнения атомов, ионов их движению к электродам. Эта сила по закону Ома равна произведению величины тока (I, А) на сопротивление ячейки (R, Ом). Омическое падение напряжения IR приводит к увеличению потенциала, необходимого для работы электрохимической ванны, или к уменьшению измеряемого потенциала гальванического элемента.
В общем случае (3):
 (3)
(3)
где  и
и  – потенциалы катода и анода.
– потенциалы катода и анода.
Следует отметить, что полного подчинения закону Ома в проводниках II рода нет. При достаточной силе тока наблюдаются отклонения вследствие электродных явлений или поляризации. Различают поляризацию:
– концентрационную (силы диффузии, электростатического притяжения и механического перемешивания не обеспечивают массопереноса реагирующего вещества к поверхности электрода или отвода от нее со скоростью, достаточной для поддержания теоретически рассчитанной по закону Ома силы тока);
– кинетическую (обусловлена малой скоростью электрохимической реакции на электроде). При этом сила тока контролируется скоростью переноса электронов, а не массопереносом реагирующего вещества.
Содержание в растворе веществ, способных электроокисляться или электровосстанавливаться на электродах, снижает их поляризацию, смещая систему к равновесному состоянию.
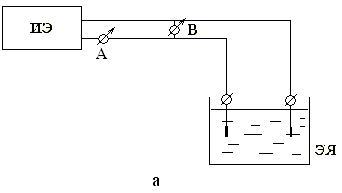
Таким образом, процессы, протекающие в электрохимической системе в значительной степени зависят от вида, концентрации компонентов межэлектродного раствора и могут быть проконтролированы с использованием измерительной схемы, упрощенно представленной на рис. 1. а.
Согласование подобных схем с IBM-совместимыми компьютерами позволяет создать виртуальные измерительные комплексы, т.е. системы сбора и обработки информации.
Аналитические сигналы со стандартных приборов (амперметры, вольтметры), имеющих самые разные выходные параметры и пределы, обрабатываются достаточно простыми устройствами. Например, в случае контроля тока может быть использована микросхема МАХ 471, содержащая несколько операционных усилителей (рис. 1. б). Диод D 5 обеспечивает индикацию измеряемого тока, что может оказаться необходимым, например, в случае снятия полных вольтамперных зависимостей. Виртуальный вольтметр может быть перепрограммирован в термокомпенсатор для потенциометрии (программа DE GRES.BAS), т.к. стабилитроны серии LM имеют значительные температурные коэффициенты напряжения (10 мВ/К). Так, рабочие диапазоны температур LM-335 от 233 до 373 К; LM-135 от 233 до 423 К (соответственно, выходные напряжения 2,23÷4,23 В). Эти параметры соответствуют интерфейсному устройству, в качестве которого целесообразно использование аналого-цифровых преобразователей (АЦП), например, на базе сдвоенного операционного усилителя LM-358 (0÷5 В), подключаемого к стандартным последовательным или параллельным портам ПК. При этом, как правило, достаточно коэффициента усиления в интервале 10÷100.
в 
| |
| Интерфейсное
устройство (АЦП, 0-3 А, 0-5 В)
| |
| б
| |

| Рис. 1. Схема измерения параметров электрохимической системы и
согласования амперметра и вольтметра с IBM-совместимыми компьютерами:
а – измерительная схема; б – схема согласования амперметра;
в – схема согласования вольтметра;
ИЭ – источник электроэнергии с регулируемым напряжением (током);
А – амперметр; В – вольтметр; ЭЯ – электрохимическая ячейка
| |
Программное обеспечение и драйверы этих и подобных устройств находятся на сервере http://www.dmk.ru или могут быть заказаны по e-mail: [email protected].
Согласно объединенному закону Фарадея, масса электроактивного
компонента раствора (m, г) может быть определена по уравнению(4):
 , (4)
, (4)
где Q – количество электричества в кулонах.
Очевидно, что данное соотношение правомерно лишь при эффективности тока, близкой к 100 %, т.е. отсутствии конкурирующих процессов.
Стабильность и количественное протекание конкретной реакции можно обеспечить созданием определенных условий ее проведения.
А. Лингейн классифицировал методы кулонометрии в соответствии с электрическими параметрами, поддерживаемыми постоянными в ходе анализа, на три группы:
а) гальваностатические, т.е. при I = const.
В этом случае потенциал рабочего (генераторного) электрода соответствует контролируемому электродному процессу лишь при количестве элекроактивного вещества, достаточном для переноса приложенного тока. Снижение концентрации изменяет потенциал до значений, допускающих протекание других процессов, поддерживающих приложенный ток. Полное количество прошедшего через раствор электричества определяется произведением величины тока на время (5):
 ; (5)
; (5)
б) потенциалостатический, т.е. при Еяч . = const.
Такой анализ при постоянстве приложенного на ячейку напряжения обеспечивает бόльшую селективность, т.к. величина Еяч. может быть достаточно малой, и при прекращении контролируемого электрохимического процесса потенциал рабочего электрода не способен сместиться до величины, достаточной для протекания побочной реакции. Особенностью этого контроля является его продолжительность, обусловленная малыми токами;
в) потенциостатический, т.е. при φэл = const.
Выбранный потенциал рабочего электрода, определяемый как ЭДС между ним и электродом сравнения, обеспечивает в данном случае не только селективность анализа, но и наиболее оптимальный (максимальный) при данных условиях ток электролиза. Необходимая величина ЭДС поддерживается путем изменения полного напряжения на ячейке Еяч, приложенного на рабочий и третий, вспомогательный, электрод. При этом происходит компенсация изменений в ходе анализа потенциалов электродов и перенапряжений.
При потенциало- и потенциостатическом контроле количество прошедшего электричества определяют интегрированием (6):
Q =  , (6)
, (6)
Вне зависимости от поддерживаемых постоянными тока или потенциалов (Еяч, φэл), кулонометрию подразделяют на прямую и косвенную (кулонометрическое титрование).
В прямой кулонометрии имеет место непосредственное электроокисление или электровосстановление определяемого вещества на электроде. При реализации прямой кулонометрии работают, как правило, в потенциостатическом режиме, поддерживая потенциал рабочего электрода на уровне, обеспечивающем предельный диффузионный ток. Гальваностатический режим в прямой кулонометрии применяют только в случаях, когда определяемое вещество находится на рабочем электроде в твердом состоянии или когда его предварительно выделяют при любом режиме на электрод в виде твердой фазы, растворяемой затем при I = const до появления скачка потенциала.
При кулонометрическом титровании в систему вносят электроактивный реагент, а продукты электролиза химически взаимодействуют с определяемым веществом. Момент завершения реакции определяемого компонента с генерируемым веществом фиксируют визуально по изменению окраски предварительно введенного индикатора или инструментально с использованием потенцио-, амперо- или фотометрии.
Наиболее распространено кулонометрическое титрование при постоянном токе. При этом выход по току (η, %) определяется по уравнению(7):
 (7)
(7)
где iоб и i – плотности тока электролиза в присутствии вспомогательного реагента и без него, соответственно, при одном и том же значении потенциала.
В качестве материала рабочих (генераторных) электродов, на которых протекает заложенная в основу кулонометрического анализа электрохимическая реакция, используют, как правило, платину (для анодных процессов) или ртуть (для катодных). Эти же материалы применяют и для изготовления вспомогательных электродов. При потенциостатическом кулонометрировании в ячейку дополнительно помещают электроды сравнения, относительно которых контролируется потенциал генераторного электрода. Для поддержания постоянства силы тока или потенциалов при протекании электрохимических или химических процессов в системе используют гальваностаты (амперостаты) или потенциостаты. Оптимально наличие универсальных устройств, обеспечивающих высокий уровень стабилизации и регулирования как по току, так и по напряжению. Лучшими являются блоки питания типа «Tesla», классифицируемые по интервалам этих величин. Для измерения полного количества электричества, прошедшего через ячейку в ходе анализа используют интегральные электронные кулонометры для измерений в диапазоне 10–5¸104 Кл при точности 0,1 % и скорости внутреннего дрейфа во время интегрирования менее 10–7 Кл/с. Наиболее распространены кулонометры, содержащие в основе:
– операционный усилитель, в цепи обратной связи имеющий конденсатор, работающий практически без утечек. По величине напряжения на конденсаторе определяется заряд, прямо пропорциональный прошедшему за определенное время току;
– преобразователь напряжения на прецизионном резисторе в частоту, измеряемую пересчетной схемой. Такая система отличается более высокими линейностью и быстродействием.
В обоих случаях кулонометры градуируются для получения отсчетов в кулонах или микрограмм-эквивалентах. В качестве первичных стандартов для прецизионных калибровок используют, как правило, химические кулонометры, например, работающие на принципе контроля растворения серебра. Получаемые на нем значения электрохимического эквивалента серебра (1,117972±0,000019 мг/Кл) и числа Фарадея (96490,0±2,4 Кл/моль) позволяют работать в диапазоне 0,015÷75 Кл со стандартным отклонением 0,096 %.
Лабораторные работы
Работа 2. Количественное определение тиосульфата натрия
Теоретические основы
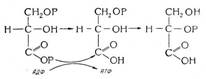
Титрование тиосульфата натрия проводят йодом, электрогенерированным на аноде из йодида калия, применяя визуальное обнаружение конечной точки с помощью крахмала. При этом протекают следующие реакции:
на аноде: 2I- - 2e- → I2
в растворе: I2 + 2e- → 2I- E0 (I2/2I-) = 0,54 B
2S2O32- - 2e- → S4O62- E0(S4O62-/2S2O32-) = 0,20 B
Эквивалентную точку определяют по появлению синего окрашивания раствора в результате образования адсорбционно комплексного соединения йода с крахмалом. Для анализа используют стандартную кулонометрическую установку, состоящую из последовательно соединенных блоков питания, магазина сопротивлений, миллиамперметра и ячейки с двумя платиновыми электродами без разделения приэлектродных пространств диафрагмой.
Методика проведения анализа
Анализируемый раствор доводят до метки в мерной колбе на100,00 мл и перемешивают. В кулонометрическую ячейку наливают цилиндром 10 мл 1М раствора KI, 2 мл 0,01 М раствора Н2SO4, 1 мл раствора крахмала (1%) и пипеткой аликвоту (10,00 мл) анализируемого раствора. Помещают в ячейку платиновые электроды, добавляю дистиллированной воды до полного покрытия их поверхности и перемешивают полученную систему в течении 1-2 минут на магнитной мешалке. Подают на ячейку электрический ток, включают секундомер (при отсутствии в цепи интегрального кулонометра) и устанавливают магазином сопротивлений рабочий ток 10,0 мА. При появлении синего окрашивания раствора, обусловленного появлением избытка молекулярного йода, электрическую цепь размыкают и одновременно останавливают секундомер. Фиксируют время электролиза (τ, с). Аналогично проводят повторное (параллельное) титрование.
Обработка экспериментальных данных
Определяют по показаниям кулонометра ИПТ-1 или рассчитывают количество электричества, затраченное на каждое титрование (Q) как произведение величины тока (А) на продолжительность анализа (сек.). Содержание тиосульфата натрия (г/л)определяют по уравнению:
 , г/л
, г/л
где М – молекулярная масса Na2S2O3 (138);
n – число электронов (1);
Val – объем аликвоты исследуемого раствора;
F – число Фарадея (96485 Кл/моль);
Находят среднее значение содержание определяемого вещества в растворе (Сср Na2S2O3, г/л).
Работа 3. Определение следовых количеств кислот, оснований и гидролизующихся солей
Теоретические основы
При электролизе водных растворов потенциалы электродов обеспечивают разложение растворителя (Н2О) с образованием ионов водорода (на аноде) и гидроксильных ионов (на катоде). Генерируемые количества ионов дозируются до очень малых величин изменением точного контролируемых параметров тока и времени.
В ходе анализа растворов различной кислотности (основности) на электродах протекают реакции:
- анодная, Н2О – 2е- → ½ О2 + 2Н+
- катодная 2Н2О +2е- → Н2 + 2ОН-
Образовавшиеся на электродах ионы титранта вступают в протолитическое взаимодействие с компонентами анализируемых систем (в растворе): Н+ + ОН- → Н2О
Избыток титрантов (Н+; ОН- - ионы) по завершении последней реакции фиксируется изменением цвета индикаторов, например гематоксилина (рТ=6,0) или фенолфталеина (рТ=9,0), содержащихся соответственно в анодной или катодной камерах.
Для исключения погрешностей, обусловленных наличием в растворах СО2, проводят предэлектролиз фонового раствора по реакции образования бикарбоната:
2Н2СО3 + 2е- → Н2 + 2НСО3-
Методика проведения анализа
Анализируемый раствор доводят до метки в мерной колбе на 100,00 мл и перемешивают. Используя универсальную индикаторную бумагу, определяют рН исследуемой системы (покраснение бумаги – рН < 7, кислая; посинение – рН > 7, основная среда). В соответствии с таблицей выбирают условия анализа.
Таблица. Условия проведения протолитической кулонометрии
| Характеристика исследуемого раствора, рН
| Рабочая камера
| Поляризация генераторного электрода
| Генерируемые ионы (титрант)
| Применяемый индикатор, рТ
| Переход окраска индикатора
|
| Кислый рН<7
| Катодная
| Отрицательная
| ОН-
| Фенолфталеин, 9,0
| б/цв ↔ слабо- розовый
|
| Нейтральный рН≈7
| Протолитическое титрование невозможно
|
| Основной рН>7
| Анодная
| Положительная
| Н+
| Гематоксилин6,0
| Желтый↔
фиолетовый
|
Завешивают в ячейку малую камеру с пористой диафрагмой и в обе камеры заливают (мерным цилиндром) по 15 мл 10% раствора сульфата калия (К2SO4; фон). В рабочую (большую) камеру добавляют 50 мл дистиллированной воды и 5-6 капель соответствующего индикатора (см. табл.). В ячейку опускают пару электродов, контролируя полярность генераторного (см. табл.) и включают перемешивание (без образования воронки в растворе). Проверяют превышение уровня раствора в малой камере на 1-2 см и полноту погружения поверхности электродов. При необходимости добавляют в соответствующую камеру Н2О дист.
При окрашивании системы в слабо-розовый (фенолфталеин) или фиолетовый (гематоксилин) цвета проводят предэлектролиз. Для этого включают ток, устанавливают его магазином сопротивлений на уровне 10,0 мА и определяют время, необходимое для изменения окраски индикатора (τпредэл., сек).
В генераторную (большую) камеру вносят пипеткой 5,00 мл исследуемого раствора и, после перемешивания в течении 1-2 минут, включают ток. При отсутствии в схеме интегрального кулонометра ИПТ-1 фиксируют время начала электролиза и магазином сопротивлений устанавливают значение тока, равное 10,0 мА. Электрогенерацию титранта (Н+, ОН- - ионы) проводят до появления соответствующей окраски раствора в генераторной камере (слабо-розовая фенолфталеина, желтая гематоксилина). Отключив источник электропитания ячейки, фиксируют показания кулонометра или время титрования (τтитр., сек). Аналогично проводят повторное (параллельное) предэлектролиз и титрование новой пробы исследуемого раствора.
Обработка экспериментальных данных
Определяют по показаниям кулонометра ИПТ-1 или рассчитывают количество электричества, затраченного на титрование (Q) как произведение величины тока (А) на продолжительность титрования (τтитр., сек). Титр исследуемого i-того раствора определяют по результатам каждого опыта, используя уравнение:
 ; г/мл
; г/мл
где М – молекулярная масса i-того вещества;
Q – затраченное количество электричество А·с;
n - основность i-того соединения;
F – число Фарадея (96485 Кл/моль);
Val – объем аликвоты исследуемого раствора.
Находят среднее значение Тi ср (г/мл).
Работа 4. Кулонометрическое определение цистина
Из всех аминокислот цистин обладает наибольшими кислотными свойствами (рК1 соон<1, рК2 соон =1,7), что позволяет проводить амперостатическое кулонометрическое титрование:



В ходе предэлектролиза фонового водного раствора сульфата калия происходит разложение молекул воды:
2 Н2О + 2 ē → Н2 + 2 ОН-.
При введении анализируемого раствора в катодную камеру цистин [НООССН(NН2) СН2S]2 восстанавливается в щелочной среде до цистеина НSCH2CH(NH2)COOH и далее разлагается на Н2S, NН3 и пировиноградную кислоту. Эти процессы проходят через ряд промежуточных стадий (более десяти), вид и последовательность которых различны при изменении концентрации основного вещества, ионной силы раствора, условий проведения анализа и др. факторов.
Методика проведения анализа. Рабочую ячейку заполняют до половины фоновым раствором (5% сульфат калия). Завешивают анодную камеру и наливают в нее из бюретки К2SО4 в количестве, обеспечивающем превышение уровня жидкости на 1-2 см по отношению к верхнему слою раствора в катодной камере. Опускают электроды (меньший по размеру - катод) до их полного погружения. В катодную камеру вводят 3-4 капли фенолфталеина, включают мешалку и электрическую цепь кулонометрической установки. Используя магазин сопротивлений, устанавливают ток в пределах 10-15 мА и проводят предэлектролиз до появления слабо-розового окрашивания фонового раствора (рН=9). Электролиз прекращают.
В катодную камеру приливают из бюретки 2,00 мл раствора цистина (окраска исчезает) и, включив электрическую цепь, определяют интегральным кулонометром количество электричества Q, затраченное на повторную регенерацию гидроксильных ионов до рН=9 (появление слабо-розового окрашивания). В ходе процесса контролируют величину тока, поддерживая ее на уровне 10-15 мА.
Повторяют титрование, вводя последовательно 2,00; 4,00; 5,00; 6,00 мл анализируемого раствора без замены фона.
Обработка экспериментальных данных. Определяют весовое содержание цистина в пробах Рi и титры растворов его (Т i) по результатам (Qi) каждого опыта, используя уравнения:
Рi =  , г.
, г.
где Q - количество электричества, затраченного на электрохимическое образование гидроксильных ионов, Кл;
М - молекулярная масса цистина (240,24);
n - число электронов, участвующих в электрохимической реакции;
F - число Фарадея (96485 Кл/моль), т. е. количество электричества (1 моль электронов), необходимое для электрохимического превращения 1 г-экв вещества.
T i =  , г/мл,
, г/мл,
где Vi – объем соответствующей пробы цистина, мл.
Проводят статистическую обработку полученных результатов и определяют погрешность метода анализа, используя соответствующие методические указания.
Вопросы для самоконтроля
1. Сущность кулонометрического анализа.
2. Понятие и физический смысл электрохимического эквивалента вещества, постоянной Фарадея.
3. Принципиальные схемы кулонометрических установок.
4. Основные характеристики элементов кулонометрических устройств.
5. Виды кулонометрии (прямая, косвенная).
6. Классификация методов кулонометрии по постоянству электрических характеристик.
7. Определение затрат электроэнергии в различных методах кулонометрии.
8. Требования к реакциям в кулонометрическом анализе.
9. Различные виды электродов в кулонометрии.
10. Обработка экспериментальных данных кулонометрического анализа.
11. Способы калибровки кулонометров.
12. Достоинства и недостатки кулонометрии.
Решение типовых задач
Задача 1. При кулонометрическом титровании пробы четырехвалентного церия объемом 5,00 мл электрогенерируемыми ионами двухвалентного железа при силе тока 24,0 мА потребовалось 100 секунд. Определить содержание (г/л) Се4+ в исследуемом растворе.
Решение. В ходе анализа протекают реакции:
а) на электроде Fe3+ + 1e- → Fe2+
б) в растворе Се4+ + 1е- → Се3+
В соответствии с объединенным законом Фарадея (4):
 , (г/л)
, (г/л)
После подстановки численных значений:
 г/л.
г/л.
Задача 2. Навеску латунного припоя 0,6578 г растворили и через полученный раствор в течении 20,0 мин пропускали ток силой 1,0 А. При анализе на катоде количественно выделилась медь по реакции (выход по току 80%):
Cu2+ +2e- → Cu0
Определить процентное содержание (А%) меди в припое.
Решение. Определим количество электричества, затраченного на анализ:
 кул (А·с)
кул (А·с)
Массу меди, выделившейся на катоде, рассчитываем по уравнению Фарадея (4):
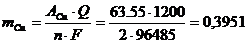 г
г
Тогда процентное содержание меди в припое (А%) равно:

Задача 3. На полное восстановление цинка из пробы 10,00 мл при силе тока 100 мА затрачено 26 мин. Определить содержание цинка (г) в пробе и молярную концентрацию его в исследуемом растворе.
Решение. Содержание цинка в пробе (г) находим по уравнению Фарадея(4):
 г.
г.
Число молей цинка в одном литре исследуемого раствора:
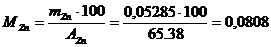 г-моль/л.
г-моль/л.
Задача 4. При кулонометрическом контроле раствора, содержащего ионы трехвалентного металла при силе тока 1,0 А затрачено 15 минут. В ходе анализа масса катода увеличилась на 0,6497 г. Какой металл содержался в исследуемом растворе, если выход по тку составил 100%?
Решение. Используя уравнение Фарадея (4), рассчитаем атомную масса количественно выделившегося на рабочем (генераторном) электроде металла:

По периодической системе (таблице) химических элементов Д. И. Менделеева определяем наличие в исследуемом растворе висмута (А.м=208,98).
ПОЛЯРОГРАФИЧЕСКИЙ АНАЛИЗ
Метод полярографии предложен для анализа чешским ученым Ярославом Гейровским (1890-1967) в 1922 г. Первый полярограф Я. Гейровский и М. Шиката сконструировали в 1925 г. Я. Гейровский руководил созданным в Праге в 1946 г. институтом полярографии. В 1959 г. за создание и развитие метода удостоен Нобелевской премии.
Основными преимуществами полярографии являются возможности одновременного качественного и количественного контроля проб, определения ряда ионов при их совместном присутствии без предварительного разделения, проведения повторных анализов одной и той же пробы (неразрушающий контроль).
Теоретические основы
1. Сущность и основные закономерности метода
Наложенное на электролитическую ячейку напряжение Е вызывает поляризацию анода, катода и расходуется на прохождение тока через раствор (1):
Еяч = φа – φк + IR (1)
При соблюдении условий:
а) R мало в присутствии индифферентного электролита (фона), т.е. падением напряжения в растворе можно пренебречь, IR " 0.
б) соотношение электродных площадей Sк << Sa, следовательно, плотности тока (и величина поляризации на электродах) iк >> ia (А/см2), причем φа = const.
Имеем  (катодная поляризация).
(катодная поляризация).
Аналогично при Sк >> Sa, iк << ia, φк = const:
 (анодная поляризация).
(анодная поляризация).
Повышение накладываемого на ячейку напряжения обуславливает увеличение поляризации микроэлектрода. В качестве индикаторного микроэлектрода обычно используют капли ртути, вытекающие из капилляра. Электродом сравнения служит слой ртути, помещенной на дно электролизера. Схема типичной полярографической установки представлена на рис.1.

Рис.1. Схема полярографирования на ртутных электродах:
1 -источник напряжения
(аккумулятор, стабилизированный
выпрямитель);
2-регулятор напряжения (реостат);
3-ячейка;
В – вольтметр, А - амперметр
Контроль величины тока, идущего через систему II позволяет судить о происходящих на микроэлектроде «процессах. Получаемые зависимости I = f (E) называются вольт-амперными кривыми или полярограммами (рис.2.1).

Рис.2. Полярограммы (2.1) электроактивного вещества при концентрациях С С С С и кривая амперометрического титрования (2.2) этого вещества при потенциале Е.
Полярограмма каждого компонента состоит из 3 участков:
о - а) участок до достижения потенциала разряда компонента, например, на катоде-участок остаточного тока (2):
Iост = Iк + Iф» 10–7 А, (2)
где Iк – конденсаторный ток, обусловленный подходом (но не разрядом) катионов к микроэлектроду и образованием двойного электрического слоя (конденсатора);
Iф – фарадеев ток, возникающий при разряде примесей, например, кислорода.
Некоторое увеличение Iост связано с возрастанием Iк, т.к. ток заряжения конденсатора совпадает по направлению с катодным током;
а - б) участок полярографической волны. При достижении потенциала восстановления ионов определяемого компонента (например, Me+n) сила тока резко возрастает вследствие разряда их на микрокатоде:
Me+n + Hg + nē " Me (Hg) (амальгама).
После падения капли она соприкасается с анодом и металл снова переходит в раствор (неразрушающий контроль):
Me(Hg) – nē " Me+n + Hg;
б - в) участок предельного тока. При достижении определенной величины тока устанавливается I» const. Это постоянство обусловлено достижением максимальной интенсивности подвода определяемых ионов к катоду за счет диффузии и уравниваем ее со скоростью разряда (3):
Iпред = Iмигр + Iдифф, (3)
где Iмигр – миграционный ток, обусловленный передвижением ионов под действием электрической силы (пропорционален градиенту электрического потенциала);
Iдифф – диффузионный ток, поддерживаемый за счет диффузии ионов к электроду (пропорционален градиенту концентрации определяемого компонента).
При наличии в растворе нескольких компонентов полярографическая волна повторяется по мере достижения потенциалов разряжения второго, третьего и т.д. ионов (полярографический спектр).
S-образный вид полярограмм свидетельствует о возможности как качественного, так и количественного анализа исследуемых растворов. При качественном анализе исходят из того, что потенциал начала восстановления (или окисления) деполяризатора не совсем точно характеризует компонент, т.к. смещается в отрицательную область при уменьшении концентрации раствора. Для определения природы компонента корректнее пользоваться значением середины полярографической волны. При этом исходят из уравнения Гейровского-Ильковича (4):
 (4)
(4)
где  и
и  – потенциал и соответствующий ему ток полуволны.
– потенциал и соответствующий ему ток полуволны.
Количественное определение концентрации искомого компонента осуществляют по значениям Iпред используя уравнение, предложенное Д. Ильковичем в 1936 г. (5):
 ; где (5)
; где (5)
n – число электронов, участвующих в реакции;
D – коеффициент диффузии, см /с;
m – масса капли ртути, г;
τ – период капания ртути, с.
или 
Для конкретного определяемого компонента (n, D постоянны) и капилляра (m×τ = const) при данной температуре (влияющей на D, m и τ) в случае обратимости процессов восстановления или окисления как ионов, так и нейтральных молекул (6)
 (6)
(6)