На завершающей стадии анализа часто используют прямое потенциометрическое измерение. Методика таких измерений проста и состоит в том, что сравнивается потенциал индикаторного электрода в растворе определяемого вещества с потенциалом этого же электрода, погруженного в стандартный раствор того же вещества. Измерив потенциал электрода для растворов известной концентрации или активности, можно построить калибровочный график, по которому затем можно найти концентрацию (активность) раствора того же вещества, измерив в тех же условиях потенциал раствора. Метод калибровки сочетает такие достоинства, как простота, экспрессность, пригодность для длительного определения.
Потенциометрическое титрование
С помощью подходящего индикаторного электрода удобно устанавливать точку эквивалентности. Этот метод особенно удобен при определении мутных и окрашенных растворов. Потенциометрический метод может быть использован для всех видов титрования: кислотно-основного, окислительно-восстановительного, комплексонометрического, осадительного.
При определении вещества потенциометрическим методом необходимо иметь в виду следующее:
1) чувствительность метода определяется правильным выбором индикаторного электрода;
2) величина скачка вблизи точки тем больше, чем выше концентрации стандартного и исследуемого растворов;
3) величина скачка также зависит от константы ионизации слабой кислоты или слабого основания и тем больше, чем больше соответствующая константа;
4) в случае титрования смесей двух или более веществ сначала оттитровывается вещество, имеющее более высокую степень ионизации, окислительно-восстановительный потенциал, величину ПР или устойчивость образующегося комплексного соединения;
5) в начале титрования титрант можно приливать большими порциями, а в близи эквивалентной точки (на это укажут большие изменения показаний прибора) стандартный раствор следует приливать одинаковыми порциями не более 0,2 мл.
Для определения конечной точки титрования можно использовать различные способы. Наиболее простой в построении графика зависимости рХ от объема приливаемого титранта (рис. 1а). Конечную точку титрования определяют как среднюю точку участка, соответствующего вертикального подъема кривой. Однако этот способ не всегда позволяет зафиксировать конечную точку титрования, особенно в случае титрования смесей веществ либо веществ, отличающихся малыми значениями соответствующего физико-химического параметра (константа нестойкости, окислительно-восстановительный потенциал и т.д.).
Другие способы расчета конечной точки состоят в определении изменения потенциала на единицу изменения объема ΔрХ/ΔV от объема титранта (рис. 1б) либо в определении точки, в которой вторая производная Δ2pX/ΔV2 от объема титранта равна нулю (рис. 1в).
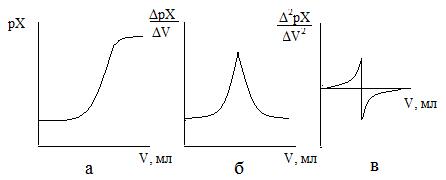
Рисунок 1. – Обработка кривых титрования
Иономеры и рН-метры. Для определения разности потенциалов используют высокоомные вольтметры заводского изготовления, шкала которых откалибрована в единицах рН и милливольтах. В число таких приборов входят ЭВ-74, рН-340, рН-510 «Эксперт – 001» и др.
В качестве электродов сравнения чаще используют хлорсеребряный электрод, в качестве индикаторных – стеклянный (для контроля рН среды), платиновый (для контроля окислительно-восстановительного потенциала), ионоселективные – серебряный (для контроля галогенидов), калиевый, кальциевый, фторидный и др.
Примеры научно-исследовательских работ
1. Прямая потенциометрия
Метод основан на измерении э.д.с. селективного электрода в нескольких стандартных растворах с известной активностью определяемого иона. С этой целью из исходного 1М раствора определяемого иона путем последовательного разведения готовят растворы с концентрацией от 0,1 до 10-5М. Измеряют э.д.с. для полученных растворов и по результатам измерений строят график зависимости Е от lg ai(pai). Такие графики, как правило, линейны, угловой коэффициент соответствует электродной функции по Нернсту (2,3 RT/nF на единицу раi). В тех же условиях определяют э.д.с. в исследуемом растворе и по графику находят соответствующую ей активность анализируемого иона.
По стандартному раствору соляной кислоты с концентрацией от 0,1 до 10-5 М построить калибровочный график Е – ра i. По заданию преподавателя определить активность ионов водорода в растворах минеральных и органических кислот при различной ионной силе раствора. Экспериментальные данные сопоставить с теоретическими расчетами, выполненными на основании уравнения Дебая-Гюккеля.
2. Определение серной и фосфорной кислот при их совместном присутствии.
Количественное определение серной и фосфорной кислот при их совместном присутствии основано на измерении рН этого раствора в процессе титрования щелочью со стеклянным электродом. Полученные кривые имеют две точки эквивалентности, определяемые по изменению хода кривых титрования. Первая точка эквивалентности соответствует нейтрализации всей серной и фосфорной кислоты до дегидрофосфата натрия.
H2SO4 + 2NaOH = Na2SO4 + 2 H2O
H3PO4 + NaOH = NaH2PO4 + H2O
Вторая точка эквивалентности соответствует нейтрализации второго иона водорода фосфорной кислоты с образованием гидрофосфата натрия.
NaH2PO4 + NaOH = Na2HPO4 + H2O
Раздельное титрование первого и второго ионов водорода фосфорной кислоты возможно в связи с большой разницей в величине констант диссоциации фосфорной кислоты и по первой и по второй ступени (рК1 = 2,12; рК2 = 7,21; рК3 = 12,36). Так как в результате титрования получается однозамещенная, а затем двухзамещенная соль фосфата натрия, то рН точек эквивалентности может быть определена по формулам:
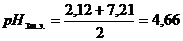
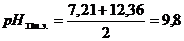
Ход определения.
Пробу полученную для анализа раствора смеси кислот (~0,5н) 10 см3 количественно переносят в мерную колбу на 100 см3 и доводят в колбе до метки водой. Пробу 20 см3 переносят в стакан емкостью 150-200 см3 и разбавляют водой до 70-100 см3. При перемешивании фиксируют начальное значение рН. Затем начинают приливать из бюретки щелочь, записывая показания прибора после прибавления первой порции по 0,5 см3. В области первого скачка титрования (рН 4-6) и второго скачка (рН 9-11) объем добавляемой порции раствора уменьшают до 0,1-0,2 см3. Титрование повторяют четыре раза. По окончании работы электроды промывают дистиллированной водой. По данным строят график зависимости ΔpH/ΔV-V.
По максимуму на кривой титрования определяют объем едкого натра (V1), затраченного на титрование серной кислоты и фосфорной кислоты по первой ступени диссоциации, и объем едкого натра (V2), затраченный на титрование фосфорной кислоты по второй ступени (рис. 2). Расчет содержания кислот в анализируемом растворе проводят по формуле:



Рисунок 2. – Кривая титрования
3. Определение смеси сильной и слабой кислоты при совместном присутствии (на примере HCl и H3BO3)
Константа диссоциации борной кислоты по первой ступени очень мала ( ). Это не дает возможности получить четкий скачек потенциала. Прибавление многоатомных спиртов (глицерин, глюкоза, пропиленгликоль и др.) приводит к образованию сложных кислот, например глицерино-борной H{BO2[C3H5(OH)5]} с Кдисс= 3·10-3. Повышение константы диссоциации способствует получению четкого скачка и возможности фиксирования эквивалентной точки.
). Это не дает возможности получить четкий скачек потенциала. Прибавление многоатомных спиртов (глицерин, глюкоза, пропиленгликоль и др.) приводит к образованию сложных кислот, например глицерино-борной H{BO2[C3H5(OH)5]} с Кдисс= 3·10-3. Повышение константы диссоциации способствует получению четкого скачка и возможности фиксирования эквивалентной точки.
Ход определения
Предварительно устанавливают значение рН раствора в конечных точках для титрования соляной и борной кислот. С этой целью в сосуд для титрования помещают 5-10 см3 0,1 н. борной кислоты и разбавляют водой так, чтобы покрыть рабочую часть электродов. Измеряют величину рН раствора борной кислоты (рН1). Затем в этот раствор вносят 10 см3 глицерина и титруют раствором гидроксида натрия порциями по 0,5 см3, измеряют величину рН раствора после каждой порции титранта. Строят график в координатах ΔpH/ΔV-V. Максимальное значение величины ΔpH/ΔV соответствует значению рН конечной точки титрования глицерино-борной кислоты (рН2).
Затем, промыв электроды, в стакан для титрования помещают заданное количество смеси HCl и H3BO3 и доливают водой, чтобы покрыть рабочую часть электродов. Титруют 0,1 н. раствором NaOH до достижения значения рН (порции титранта вблизи этого значения уменьшают до 0,1 см3) и записывают объем титранта V1. Затем в раствор вводят глицерин (10-20 см3) и титруют до достижения значения рН2. Записывают объем титранта V2. Разность (V2-V1) дает объем титранта, эквивалентный содержанию борной кислоты. Содержание HCl и H3BO3 рассчитывают по формуле:

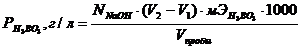
4. Определение Fe2+ титрованием бихроматом калия
Определение основано на окислительно-восстановительной реакции
 6Fe2+ + Cr2O72- + 14H+ 6Fe3+ + 2 Cr3+ + 7H2O
6Fe2+ + Cr2O72- + 14H+ 6Fe3+ + 2 Cr3+ + 7H2O
до достижения эквивалентной точки:
 ;
;
после эквивалентной точки:
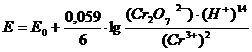

В качестве индикаторного используют платиновый электрод.
Ход определения
В ячейку для титрования помещают 5-20 см3 испытуемого раствора и 0,1 н серной кислоты в количестве, чтобы закрывать рабочую поверхность электродов. Титрант прибавляют порциями по 0,5 см3, вблизи точки эквивалентности титрант прибавляют по 0,1-0,2 см3. Строят график в координатах Е-Vтитранта, концентрацию Fe2+ рассчитывают по формуле:
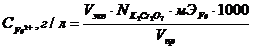 .
.
5. Определение Fe3+ комплексонометрическим методом
В кислой среде (рН=3-4) Fe3+ образует с комплексоном III (H2R2-) прочное комплексное соединение (FeR-), вследствие чего потенциал платинного электрода понижается. Как только все ионы Fe3+ окажутся связанными в комплекс, наступит резкий скачек потенциала из-за практического исчезновения окисленной формы Fe3+ системы Fe3+/ Fe2+. Дальнейшее добавление титранта не изменяет потенциал электрода.
Ход определения
В стакан для титрования помещают 5-20 см3 раствора определяемого вещества, 50 см3 дистиллированной воды и по каплям 10-процентный раствор ацетата аммония (или натрия) до тех пор пока раствор не окрасится в желто-оранжевый цвет (гидролиз соли трехвалентного железа). Добавляют 5-10 капель 1% раствора соли Мора (не содержащей Fe2+) и титруют раствором комплексона III. Следует иметь в виду, что вблизи эквивалентной точки потенциал устанавливается медленно. Строят график зависимости Е-Vтитранта. Содержание Fe3+ рассчитывают по формуле:
 .
.
Вопросы для самопроверки
1. На чем основаны потенциометрические методы анализа?
2. Какая зависимость выражается уравнением Нернста?
3. Каковы функции индикаторных электродов и электродов сравнения?
4. Какой индикаторный электрод может быть использован для измерения рН-среды?
5. Каково устройство стеклянного электрода и электрода с жидкой мембраной?
6. Типы ионоселективных электродов.
7. В каких координатах строят кривые потенциометрического титрования?
8. Достоинства и недостатки потенциометрических методов анализа.
9. Приведите пару соответствующих электродов для: а) кислотно-основного титрования; б) окислительно-восстановительного; в) осадительного; г) комплексонометрического.
10. Какие факторы влияют на скачек потенциала, рН?
Решение типовых задач
1. В стандартных растворах CaCl2 c различной концентрацией ионов Са2+ измерили электродный потенциал Са2+-селективным электродом относительно хлорсеребряного:
| С(Са2+), моль/дм3
| 1·10-1
| 1·10-2
| 1·10-3
| 1·10-4
|
| Е, мВ
| 100,0
| 46,0
| -7,0
| -60,0
|
Навеску CaCl2 (m= 0,5000 г) поместили в мерную колбу, емкостью 200 см3, растворили и довели до метки. Электродный потенциал Са2+-селективного электрода (Ех) в полученном растворе составил 34,0 мВ. Вычислить массовую долю кальция в образце.
Решение: по результатам измерения электродного потенциала Са2+-селективного электрода в стандартных растворах CaCl2 строят калибровочный график в координатах Е-рС (Са2+) (рис. 3). По графику находят, что при Ех = 34,0 мВ рС (Са2+) =2,25 и С (Са2+) = 5,6·10-3 моль/дм3.
Масса Са2+ в растворе:
m (Ca2+) = C (Ca2+)·V·A,
где V – объем приготовленного раствора, см3;
А – атомная масса Са2+, г/атом.
m (Ca2+) = 5,6·10-3·200·40 = 44,8 мг
Массовая доля Са2+ в анализируемой соли CaCl2:

При расчетах с применением графика в стандартные и исследуемые растворы вводят одинаковое количество индифферентного электролита для поддержания постоянной ионной силы растворов.

Рисунок 3. – Калибровочный график
2. Анализируемый раствор метиламина – СН3NH2 объемом 20,00 см3 разбавили в мерной колбе до 100,00 см3. Пробу полученного раствора 10,00 см3 оттитровали потенциометрически 0,1000 н HCl со стеклянным электродом. Построить кривую титрования в координатах рН-V и определить концентрацию метиламина по следующим данным:
| VHCl, см3
| 12,0
| 15,0
| 17,0
| 17,5
| 17,8
| 18,0
| 18,2
| 18,5
| 19,0
|
| рН
| 10,36
| 9,96
| 9,43
| 9,17
| 8,28
| 5,99
| 3,28
| 2,84
| 2,58
|
Строим кривую титрования: из середины вертикального участка опускаем перпендикуляр. Эквивалентный объем равен 18,0 см3. При реакции метиламина с HCl протекает реакция СН3NH2 + HCl = СН3NH3Cl. Поэтому эквивалент метиламина равен М/1. Содержание СН3NH2 рассчитываем по формуле:

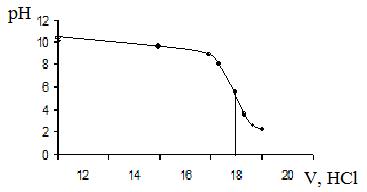
Рисунок 4. – Кривая титрования
2.2. КУЛОНОМЕТРИЧЕСКИЙ АНАЛИЗ
Основные принципы и технические основы кулонометрического анализа были определены Н. Сандом в 1907 г. Практическое использование кулонометрии долгое время сдерживало отсутствие специального оборудования. Широкое применение началось лишь с 1942 г., после создания А. Хиклингом устройства для автоматического контроля потенциала, названного им потенциостатом. Первая обобщающая монография «Кулонометрический анализ» была опубликована К. Абрешом и И. Классеном в 1961 г. Кулонометрия – единственный количественный метод анализа, не использующий зависимость свойства исследуемой системы от концентрации определяемого вещества, т.к. основан на контроле непосредственно числа электронов, участвующих в реакции. Этим обусловлены высокая чувствительность и низкий предел определяемых концентраций (от 10–8 до 10–9 М); применимость как к концентрированным, так и разбавленным растворам; отсутствие проблем, связанных с приготовлением, хранением и стандартизацией титрантов, возможность проведения анализа малыми их количествами, генерируемыми непосредственно в зоне реакции.