Переход от газовой фазы к жидкой фазе коренным образом может менять кислотно-основные характеристики кислот и оснований Бренстеда-Лаури. Первоначально рассмотрим различие в термодинамических параметрах при диссоциации кислот Бренстеда-Лаури в газовой и жидкой фазах. На рис. 4.3 приведена диаграмма изменения энтальпии при диссоциации кислоты НА в газовой и жидкой фазах.
Как следует из рис. 4.3, при переходе от газовой фазы к жидкой фазе появляются дополнительные энтальпийные составляющие, влияющие на суммарную энтальпию диссоциации (DНр-р) кислоты НА в растворе. Энтальпия недиссоциированной кислоты НА понижается вследствие ее сольватации на величину DНсольв.(НА), а энтальпия ионов (Н+ и А‾) понижается на величину DНсольв.(Н+, А‾).

Рис.4.3. Диаграмма изменения энтальпии при диссоциации кислоты НА в газовой фазе (г) и растворе (р-р).
Энтальпия диссоциации кислоты НА в газовой фазе и в растворе связаны друг с другом выражением:
DНр-р = DНг +[DНсольв.(Н+, А‾) - DНсольв.(НА)].
Исходя из точно таких же соображений можно построить диаграмму изменения свободной энергии в ходе диссоциации кислоты НА в жидкой фазе. В соответствие с этим свободная энергия диссоциации кислоты НА в растворе будет описываться уравнением:
DGр-р = DGг +[DGсольв.(Н+, А‾) - DGсольв.(НА)].
Таким образом, свободные энергии, энтальпии диссоциации кислоты НА в растворе отличаются от свободных энергий, энтальпий диссоциации кислоты НА в газовой фазе на разницу в величинах свободных энергий, энтальпий сольватации ионов .(Н+, А‾) и недиссоциированной формы кислоты (НА). Все факторы, увеличивающие силу кислот в газовой фазе, будут способствовать увеличению силы кислоты и в растворе. Однако на силу кислоты в растворе существенное дополнительное влияние оказывают сольватационные эффекты.
На рис. 4.4 приведена диаграмма изменения энтальпии при протонировании основания В в газовой и жидкой фазах.

Рис.4.4. Диаграмма изменения энтальпии при протонировании основания В в газовой фазе (г) и в растворе (р-р).
Точно такую же диаграмму можно получить для изменений свободных энергий при протонировании основания В. Из диаграммы, изображенной на рис. 4.4 следует, что величина энтальпии взаимодействия основания В с протоном в растворе равна:
DНр-р = DНг + [DHсольв.(BH+) - DHсольв.(B,H+)].
Подобное же выражение получается для свободной энергии Гиббса:
DGр-р = DGг + [DGсольв.(BH+) - DGсольв.(B,H+)].
Для количественной характеристики кислотности соединений в
разбавленных растворах используют их константы диссоциации в воде. Кислоты диссоциируют в воде в соответствии со схемой:
НА + Н2О  Н3О+ + А‾.
Н3О+ + А‾.
Константа равновесия этого процесса Ка (а –acid) равна:

Поскольку в разбавленных растворах концентрация воды практически не меняется, то часто ее концентрацию упускают. С учетом этого обстоятельства константа диссоциации кислоты вычисляется в соответствии с выражением:
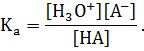
Практически при таком подходе экспериментально определяют произведение константы диссоциации на концентрацию воды. Чем больше константа диссоциации кислоты, тем сильнее кислота.
Часто для характеристики силы кислот используют не сами константы диссоциации, а их отрицательные логарифмы, которые обозначают символом рКа:
рКа = -lgKa.
Удобство этой величины измерения силы кислот заключается в том, что она прямо пропорциональна свободной энергии диссоциации:
pKa = DGдисс./2.303RT.
Чем меньше величина pKa (отрицательные числа), тем сильнее диссоциирована кислота.
Для количественной характеристики силы оснований в растворах используют константы равновесия реакций протонирования оснований в воде КВ(B – base), которые называются константами основности:
B + HOH BH+ + OH‾,

Поскольку концентрация воды в разбавленных растворах остается практически неизменной, то, как и в предыдущем случае, при вычислении констант основности концентрацией воды пренебрегают, и вычисления ведут в соответствии с выражением:

Чем сильнее основание, тем больше константа основности. Часто для характеристики силы оснований используют не непосредственно сами константы основности, а их отрицательные логарифмы, которые обозначаются символом рКВ:
pKB = -lgKB.
В этой шкале чем сильнее основание, тем меньше величина pKB. Процесс взаимодействия основания В с водой обратим. В результате этого взаимодействия основание В превращается в кислоту ВН+. Эта кислота получила название сопряженной с основанием В кислоты. Вода, которая играла в этом процессе роль кислоты, превратилась в основание – анион гидроксила. Этот анион является основанием, сопряженным с кислотой – водой. Кислотно-основные взаимодействия сопровождаются образованием новых (сопряженных) кислот и оснований. В ряде случаев для характеристики силы оснований используют не величины констант основности, а величины констант диссоциации сопряженных кислот КВН+:
 + H2O B + H3O+,
+ H2O B + H3O+,

При использовании этой шкалы более сильным основанием соответствуют меньшие величины констант КВН+. Часто используют не непосредственно эти константы нестойкости, а их отрицательные логарифмы – величины рКВН+:
рКВН+ = - lgКВН+.
Легко видеть, что произведение величин KB и КВН+ равно ионному произведению воды, которое при 25ºС равно 10-14. Отсюда следует, что:
pKB +рКВН+ = 14.
Последнее выражение позволяет легко переходить от одной шкалы измерения силы оснований к другой.
Из температурный зависимости констант диссоциации кислот Бренстеда-Лаури легко определить их термодинамические параметры (энтальпии, энтропии). Сопоставление термодинамических параметров диссоциации кислот в газовой фазе и в растворе привело к необычным результатам.
В таблице 4.5 приведены данные работы, в которой были получены термодинамические параметры диссоциации ряда карбоновых кислот в газовой фазе.
Таблица 4.5. Термодинамические параметры диссоциации замещенных уксусных кислот в газовой фазе
| Кислота
| DG(300K), ккал/моль
| DН, ккал/моль
| DS, кал/К×моль
|
| СН3СООН
| 341.5
| 348.5
| 23.3
|
| FCH2COOH
| 331.6
| 338.0
| 21.3
|
| F2CHCOOH
| 323.8
| 330.8
| 23.3
|
| F3CCOOH
| 317.4
| 324.4
| 23.3
|
Как следует из данных таблицы 4.5, энтропии диссоциации рассматриваемых кислот практически не меняются. С введением атомов фтора и с увеличением их количества происходит уменьшение энтальпий диссоциации. Это полностью соответствует имеющимся теоретическим представлениям. Фтор является самым электроотрицательным элементом и, поэтому, является заместителем с сильным –I-эффектом. Введение атома фтора в качестве заместителя в молекулу уксусной кислоты приводит к поляризации связи О-Н. Эта полярязация усиливается с увеличением количества атомов фтора в молекуле кислоты. Поэтому с введением атома фтора в молекулу уксусной кислоты, увеличением количества атомов фтора облегчается процесс образования тесных ионных пар. Атомы фтора, вследствие своего –I-эффекта уменьшают отрицательный заряд на атоме кислорода анионного остатка в тесной ионной паре. Отсюда с введением атома фтора в молекулу уксусной кислоты, увеличением количества атомов фтора облегчается процесс разъединения протона и аниона кислотного остатка. Отметим, ход наших рассуждений предполагал, что обсуждаемые эффекты будут изменять потенциальную энергию системы, т.е. энтальпию. Из данных таблицы 4.5. следует, что относительные изменения в свободных энергиях диссоциации полностью соответствуют изменениям в энтальпиях диссоциации.
В таблице 4.6 приведены термодинамические параметры диссоциации этих же кислот в воде.
Таблица 4.6. Термодинамические параметры диссоциации замещенных уксусных кислот в воде при 25°С
| Кислота
| DG, ккал/моль
| DН, ккал/моль
| DS, кал/К×моль
|
| СН3СООН
| 6.6
| 0.0
| -22
|
| FCH2COOH
| 3.5
| -1.0
| -15
|
| F2CHCOOH
| 1.8
| 0.0
| -6.0
|
| F3CCOOH
| 0.3
| 0.0
| -1.0
|
Данные таблицы 4.6 поразительны! Энтальпии диссоциации замещенных уксусных кислот не меняются и они, за одним исключением, равны нулю. Различие в энтальпиях диссоциации данных кислот, которые были в газовой фазе, полностью нивелированы сольватационными эффектами. За изменение свободных энергий диссоциации в ряду рассматриваемых кислот ответственен энтропийный член. Однако энтропии диссоциации, которые обусловлены изменением форм движения в ходе процесса, отрицательны! Казалось бы, что этого не может быть. В ходе диссоциации из одной частицы образуется две. Это приводит к тому, что продукты диссоциации имеют по сравнению с исходной кислотой, 3 дополнительных поступательных степеней движения. Это должно было привести к возрастанию энтропии системы, и энтропия диссоциации должна быть положительной. В газовой фазе, действительно, наблюдается именно это явление (таблица 4.5). Возникает ощущение, что теоретические представления, которые использовались выше, и которые лежат в основе теоретических основ органической химии, совершенно неприменимы к реакциям в жидкой фазе. Возникает вопрос, как понять, интерпретировать приведенные в таблице 4.6 данные?
В рассматриваемом случае мы встретились с особенностью процесса гидратации ионов, образующихся в ходе диссоциации кислоты. В электростатическом поле ионов удерживаются множество полярных молекул воды. Этот процесс сопровождается также образованием водородных связей между ионами и молекулами воды. О энтальпии этих взаимодействий можно судить по тому, что они не могут быть меньше энтальпии газофазной диссоциации. Молекулы воды в гидратных оболочках теряют способность к свободному поступательному, вращательному движению. Поэтому процесс образования гидратных (сольватных) оболочек сопровождается понижением энтропии системы. Явление уменьшения подвижности молекул растворителя за счет их кулоновского взаимодействия с частицами растворенного вещества получило название электрострикции. Вследствие электрострикции и происходит понижение энтропии в процессе диссоциации кислот в воде.
Чем меньше геометрический размер иона, на котором делокализован заряд (в данном случае, замещенного ацетат-аниона), тем сильнее проявляется явление электрострикции. С увеличением количества атомов фтора в замещенном анионе уксусной кислоты (таблица 4.6), возрастает геометрический размер сферы, на которой делокализован отрицательный заряд. Это приводит к ослаблению взаимодействий полярных молекул воды с этим анионом. Молекулы воды в меньшей степени теряют свою подвижность. Поэтому при переходе от уксусной кислоты к трифторуксусной идет возрастание энтропии диссоциации.
Рассмотренное явление носит общий характер. Явление электрострикции более значимо при использовании полярных растворителей, и оно менее существенно при применении неполярных растворителей. Из полученных данных следует, что сильными в растворах могут быть те кислоты, отрицательный заряд в анионе которых делокализован на большой группе атомов.
Величины рКа и рКВ в настоящее время приводятся во множестве различных справочниках, базах данных.
В таблицах 4.7 и 4.8 приведены величины рКа и рКВ некоторых кислот и оснований.
Как следует из данных таблицы 4.7, в ряду галоидводородов наблюдается та же тенденция возрастания силы кислот, что и газовой фазе. Факторы, определяющие газофазную кислотность данных соединений, играют определяющую роль и в жидкой фазе.
На существенное влияние сольватационных эффектов на кислотно-основные свойства соединений указывают следующие факты. Так, например, анилин в газовой фазе является более основным, чем аммиак. В жидкой фазе наблюдается противоположное явление (таблица 4.8.). Объясняется данное явление тем, что ароматическое ядро в анилиниевом катионе обуславливает
Таблица 4.7. ВеличинырКа ряда кислот в воде при 25°С
| Соединение
| рКа
| Соединение
| рКа
|
| NH3
|
| HJ
| -11
|
| H2O
| 15.7
| HCOOH
| 3.8
|
| H3BO3
| 9.2
| CH3COOH
| 4.8
|
| H2S
| 7.0
| C6H5COOH
| 4.2
|
| H2CO3
| 6.4
| CH3OH
| 15.5
|
| HF
| 3.2
| HOCH2CH2OH
| 15.1
|
| HNO2
| 3.4
| CF3CH2OH
| 12.4
|
| H3PO4
| 2.1
| CH2=O
| 13.7
|
| HNO3
| -1.6
| CH3CHO
| 14.5
|
| H2SO4
| -3.0
| C6H5OH
| 9.98
|
| HCl
| -7.0
| C6H5SH
| 6.5
|
| HBr
| -9.0
| C6H5NH2
|
|
Таблица 4.8. Величины рКВН+ ряда оснований в воде при 25°С
| Соединение
| рКВН+
| Соединение
| рКВН+
|
| NH3
| 9.27
| CH3C(O)NH2
| -0.51
|
| CH3NH2
| 10.62
| (H2N)2C=O
| 0.31
|
| (CH3)2NH
| 10.77
| (C2H5)2O
| -3.59
|
| (CH3)3N
| 9.80
| CH3C(O)CH3
| -7.2
|
| CH3C(NH)NH2
| 12.40
| C6H5NH2
| 4.58
|
| (H2N)2C=NH
| 13.6
| (C6H5)2NH
| 0.9
|
его гидрофобность. Поэтому сольватация такого катиона молекулами воды происходит в меньшей степени, чем сольватация аммонийного катиона. Данное обстоятельство оказывается решающим в определении относительной основности аммиака и анилина в водной среде. Отметим, что трифениламин, который в газовой фазе является более основным чем анилин, не протонируется в концентрированной серной кислоте.