Описаны клинические проявления ПМДД у лиц женского пола, которые являются носительницами мутации в гене дистрофина в гетерозиготном состоянии. Клинические признаки могут появиться в различные возрастные периоды, но чаще провоцируются гормональными перестройками в организме женщины (начало менструаций, беременность, климакс). Появление клинических симптомов может быть обусловлено двумя причинами: 1) наличие полной или мозаичной форм синдрома Шерешевского-Тернера; 2) феноменом несбалансированной лайонизации. На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
Частота встречаемости: Мышечная дистрофия Дюшенна (МДД): 1:2500-4000 новорожденных мальчиков. Частота МДБ (Беккера) составляет 1 на 20000 мальчиков.
Синдром Хантера
 главная / медицинский справочник болезней / детские болезни
главная / медицинский справочник болезней / детские болезни
Синдром Хантера - наследственное заболевание обмена веществ с Х-сцепленным рецессивным типом наследования, характеризующееся дефицитом лизосомального фермента идуронат-2-сульфатазы и накоплением мукополисахаридов в тканях. При синдроме Хантера отмечается задержка роста, макроцефалия, деформация костно-суставного аппарата, поражение кожи, сердечно-сосудистой и дыхательной системы, гепатоспленомегалия, нарушение слуха, умственная отсталость. С целью диагностики синдрома Хантера проводится консультация генетика, определение экскреции гликозаминогликанов, рентгенография костей и суставов. Пациентам с синдромом Хантера показана пожизненная ферментозамещающая терапия препаратом элапраза.
Синдром Хантера (мукополисахаридоз II типа) - редкое генетическое заболевание, сцепленное с Х-хромосомой, при котором вследствие ферментной недостаточности происходит неполное разрушение и накопление кислых мукополисахаридов (гликозаминогликанов) в различных тканях. Частота рождения детей с синдромом Хантера составляет примерно 1:100000-150000 новорожденных. В настоящее время в мире насчитывается не более 2000 больных синдром Хантера; по России официальная статистика отсутствует. Синдром Хантера относится к группе орфанных заболеваний, лечение которых, согласно действующему законодательству, должно осуществляться за счет средств федерального и регионального бюджета.
Причины синдрома Хантера
Развитие синдрома Хантера связано с мутацией гена идуронатсульфатазы (IDS), кодирующего лизосомный фермент идуронат-2-сульфатазу. Ген IDS картирован в локусе Xq28 на длинном плече Х-хромосомы; в настоящее время известно более 150 различных его мутаций (точечные мутации, мелкие и крупные делеции, вставки, перестройки и пр.).
В силу сцепленности наследования с Х-хромосомой, синдромом Хантера, как правило, страдают исключительно мальчики (XY). Гетерозиготные женщины в подавляющем большинстве случаев являются носителями мукополисахаридоза II типа без клинических проявлений, однако описаны несколько случаев синдрома Хантера у девочек, связанных с новой мутацией или инактивацией второй, нормальной Х-хромосомы.
Мутации в гене IDS сопровождаются дефицитом или отсутствием фермента iduronate-2-sulfatase (I2S), неполным расщеплением и накоплением в лизосомах клеток практически всех тканей и органов гликозаминогликанов (ГАГ) - дерматансульфата и гепарансульфата.
Классификация синдрома Хантера
Степень выраженности клинических проявлений синдрома Хантера (среднетяжелая, тяжелая) зависит от типа мутации в гене IDS. Так, крупные делеции и сложные перестройки, как правило, обусловливают развитие тяжелых форм синдрома Хантера. Для тяжелой степени (мукополисахаридоза типа IIА) характерно ранее развитие клинических симптомов (преимущественно в возрасте 18-36 месяцев), быстро прогрессирующее течение, выраженная умственная отсталость, полиорганное поражение. Пациенты с тяжелой формой синдрома Хантера погибают на втором десятилетии жизни.
Среднетяжелая форма (мукополисахаридоз типа IIВ) составляет примерно 1/3 всех случаев патологии. Клинические проявления синдрома Хантера обычно возникают у детей в 3-8 (иногда 10-13) лет; интеллект обычно сохранен; продолжительность жизни при благоприятных условиях может достигать 50-60 лет. Пациенты с легкой формой синдрома Хантера могут успешно реализовать себя в профессиональной сфере и иметь здоровое потомство.
Симптомы синдрома Хантера
На момент рождения дети с синдромом Хантера выглядят клинически здоровыми. Основная симптоматика развивается в среднем в 2-4 года. До этого возраста проявления заболевания неспецифичны: у детей могут отмечаться повторные риниты, шумное дыхание, пупочные и паховые грыжи, гидроцеле.
Ранним характерным признаком синдрома Хантера служит постепенное изменение внешности ребенка: черты лица становятся грубыми (гаргоилизм); язык, губы и ноздри – большими; кожа – толстой. Облик больного с синдромом Хантера дополняется низкорослостью, увеличением размеров головы (макроцефалией), короткой шеей, аномалиями зубных рядов (редкими зубами). Дети с синдромом Хантера внешне очень похожи друг на друга и напоминают братьев.
Другие ранние признаки мукополисахаридоза II типа включают хриплый низкий голос, увеличение живота,гепатоспленомегалию. Дети с синдромом Хантера склонны к частой заболеваемости ОРВИ, отитами,ларингитом, трахеитом, пневмониями; нередко у них отмечается обструктивное апноэ сна, хроническая диарея. Следствием отложения гликозаминогликанов и липидов в дерме служит возникновение узелково-папулезных высыпаний на коже плеч, бедер, лопаток. Возможно появление «монгольских пятен» в пояснично-крестцовой области, гипертрихоза.
Уже в дошкольном возрасте у детей с синдромом Хантера появляются признаки поражения опорно-двигательного аппарата, выражающиеся в тугоподвижности суставов, развитии кифосколиоза, деформации кистей по типу «когтистой лапы». Движения становятся затруднительными, походка неуклюжей; к юношескому возрасту больные с синдромом Хантера нередко делаются беспомощными инвалидами, прикованными к постели.
Неврологические нарушения, возникающие при синдроме Хантера, включают синдром гипервозбудимости,судороги, сообщающуюся гидроцефалию, спастическую параплегию, задержку речевого развития, прогрессирующую тугоухость. Со стороны зрительной системы обнаруживается помутнение роговицы, атипичный пигментный ретинит. К наиболее поздним признакам мукополисахаридоза II типа относятся кардиологические нарушения: появление шумов в сердце, приобретенные пороки сердца (чаще митральная недостаточность), кардиомиопатия и др.
У ребенка могут наблюдаться изменения психики: обидчивость, агрессивность. В зависимости от типа синдрома Хантера интеллектуальный дефект может быть небольшим или значительно выраженным.
Летальный исход у больных с синдромом Хантера обычно наступает от прогрессирующей сердечной илилегочной недостаточности.
Диагностика синдрома Хантера
В практике педиатра синдром Хантера встречается исключительно редко. Тем не менее, мукополисахаридоз II типа может быть заподозрен на основании клинических признаков (внешних изменений, манифестации заболевания на 2-4 году жизни, прогредиентного течения и др.). Для подтверждения диагноза дети нуждаются в консультации генетика, анализе клинико-генеалогических данных, проведении молекулярно-генетических исследований.
Важным биохимическим маркером синдрома Хантера служит повышение экскреции мукополисахаридов (дерматансульфата, гепарансульфата) с мочой, дефицит фермента идуронат-2-сульфатазы. Рентгенологические исследования костей черепа, суставов, трубчатых костей, позвоночника демонстрируют дизостоз, остеоартрит, множественные изменения позвонков.
Для обнаружения морфофункциональных изменений со стороны внутренних органов и ЦНС проводится УЗИ органов брюшной полости, ЭКГ, ЭхоКГ, электроэнцефалография, МРТ головного мозга. Морфологическое исследование тканей, полученных в результате биопсии кожи, печени, миокарда и пр., выявляет однотипные изменения - клетки, заполненные гликолипидами.
В семьях с известным генотипом и высоким риском рождения ребенка с синдромом Хантера может проводиться инвазивная пренатальная диагностика – биопсия ворсин хориона, амниоцентез или кордоцентез с определением активности идуронат-2-сульфатазы в полученном материале.
Дифференциальную диагностику синдрома Хантера следует проводить с другими формами мукополисахаридозов (прежде всего, синдромом Гурлера), а также с другими лизосомными болезнями накопления.
Лечение синдрома Хантера
Детям с синдрома Хантера необходимо пожизненное проведение ферментозаместительной терапии. На сегодняшний день единственным препаратом, зарегистрированным в США, Европе и России для лечения больных с синдромом Хантера, является Элапраза (идурсульфаза). Пациенты с мукополисахаридозом II типа должны получать Элапразу еженедельно путем внутривенного капельного введения в дозе 0,5 мг/кг массы тела.
Кроме патогенетического лечения, детям с синдромом Хантера проводятся регулярные медикаментозные курсы симптоматической и корригирующей терапии гепатопротекторами, витаминами, антиоксидантами, цитопротекторами. В комплексную терапию синдрома Хантера целесообразно включать ЛФК, физиотерапию(электрофорез лидазы на суставы, парафиновые аппликации, магнитотерапию, лазеропунктуру), занятия с дефектологом и логопедом.
Прогноз и профилактика синдрома Хантера
Синдром Хантера типа В имеет более благоприятное течение и прогноз; при своевременном и регулярном лечении продолжительность жизни больных может достигать 50-60 лет. При тяжелых формах мукополисахаридоза II типа пациенты обычно погибают до 20 лет от сердечно-сосудистой недостаточности. На сегодняшний день серьезную проблему для больных с синдромом Хантера представляет своевременное получение жизненно необходимого препарата Элапраза из-за его высокой стоимости.
Дети с синдромом Хантера нуждаются в наблюдении различных специалистов: детского генетика, педиатра,детского кардиолога, детского невролога, эпилептолога, детского офтальмолога, детского отоларинголога,детского хирурга, детского ортопеда.
Основным методом профилактики синдрома Хантера является медико-генетическое консультирование супружеских пар, имеющих вероятность рождения больного ребенка, а также проведение дородовой диагностики. Следует знать, что у больных с синдромом Хантера рождаются здоровые сыновья, а дочери выступают облигантыми носителями мутантного гена.
СИНДРОМ ЛЕША-НИХАНА
Синонимы
Ювенильная гиперурикемия, первичная X-сцепленная гиперурикемия
Частота встречаемости
1:300000.
Причины
Генетически обусловленный недостаток фермента HGPRT (гипоксантингуанинфосфорибозилтрансфераза). Вследствие отсутствия фермента происходит накопление нерасщепленных продуктов обмена, в частности, мочевой кислоты, в различных клетках организма.
Факторы риска: наличие синдрома Леша-Нихена в предыдущих поколениях.
Клиника
Условно симптомы можно разделить на три основные группы:
ÞСо стороны ЦНС:
+судорожный синдром, гиперкинезы;
+повышенный мышечный тонус;
+выраженное отставание в развитии;
+частая рвота без связи с другими причинами;
+нарушения речи (нечеткая, невнятная речь);
+м.б. эпилептические припадки;
+м.б. параличи обеих верхних или нижних конечностей (параплегии);
+отставание в умственном развитии.
Þ Симптомы, связанные с обменными нарушениями:
+полидипсия;
+полиурия;
+камни в почках;
+артриты, чаще мелких суставов стоп;
+скопления мочевой кислоты в мочках ушей (тофусы);
+появление оранжевых кристаллов (кристаллов мочевой кислоты) в моче.
Þ Поведенческие расстройства:
+необычно беспокойное поведение ребенка;
+эмоциональная лабильность (быстрая смена настроения по незначительным причинам) с преобладанием агрессивности;
+после появления зубов дети склонны к самоповреждению (укусы губ, пальцев рук, расчесы на коже) – характерный признак данного заболевания.
Þ Дополнительным симптомом может служить отставание в росте ребенка по сравнению со сверстниками.
Диагностика
Диагноз ставится по трём основным клиническим элементам:
+повышенная продукция мочевой кислоты,
+неврологическая дисфункция,
+когнитивные и поведенческие нарушения,
o решающее значение имеет генетическое исследование.
План обследования таких пациентов:
- ОАМ и КАК;
- БАК с определением уровня мочевой кислоты;
- УЗИ почек;
- консультация детского невролога, ревматолога, медицинского генетика.
Довольно сложно поставить диагноз на ранней стадии, когда эти признаки не так очевидны. Подозрения могут возникнуть из-за задержки развития, сопровождающейся гиперурикемией. Зачастую подозрения на синдром Лёша — Нихена возникают с появлением наносимых самому себе ранений у больного. Однако самотравмирующее поведение встречается и в других патологических состояниях, таких как неспецифическая умственная отсталость, аутизм, синдром Туретта, синдром Корнелии де Ланж, синдром Ретта, синдром Райли — Дея, нейрокантоцитоз, наследственные нейропатии первого типа и некоторые психиатрические заболевания.
Лечение
+Снижение уровня мочевой кислоты (ЛС, снижающие уровень мочевой кислоты).
+Удаление камней в почках медикаментозными и хирургическими методиками.
+Применение НПВС при воспалении суставов.
+Симптоматическое лечение неврологических расстройств в зависимости от преобладания того или иного симптома.
Прогноз
Т.к. течение прогрессивное и ведет к ХПН, то прогноз неблагоприятный. В зависимости от выраженности нарушений продолжительность жизни больных различна, как правило, не более 30 лет.
| Синдром умственной отсталости с ломкой Х-хромосомой
|
Синоним: синдром Мартина-Белла. Ранее эту болезнь называли синдромом олигофрении с маркерной Х-хромосомой. Синдром Мартина-Белла - одна из наиболее часто встречающихся (после болезни Дауна) форм умственной отсталости. Популяционная частота заболевания составляет 1:2000-1:5000 всех живорождённых. Больных мальчиков в 2-3 раза больше, чем девочек, мальчики болеют тяжелее.
Внешность больных неспецифична, но определённое диагностическое значение имеют удлинённое лицо, высокий выступающий лоб, макро- и долихоцефалия, гипоплазированная средняя часть лица (особенно в сравнении с выступающими и часто увеличенными щеками), выступающий подбородок. Нёбо обычно дугообразное, губы толстые, нижняя губа часто вывернута. Отмечается также увеличенный размер оттопыренных ушных раковин. Обращают на себя внимание большие кисти и стопы. Одно из типичных проявлений синдрома - макроорхидизм. При рождении у детей яички нормальных размеров, но с наступлением пубертатного периода существенно увеличиваются. Увеличение размеров гонад происходит за счёт избыточного роста соединительной ткани и накопления жидкости, но половая активность таких больных минимальна, способны к половой жизни единицы.
Могут отмечаться повышенная растяжимость кожных покровов, слабость связочного аппарата суставов (что приводит к самопроизвольными вывихам, обычно пальцев кисти), плоскостопие; у многих больных формируется пролапс митрального клапана. Все эти симптомы обусловлены врождённой дисплазией соединительной ткани.
Умственная отсталость, типичная для больных с синдромом Мартина-Белла, как правило, оценивается как умеренная или выраженная, хотя у 10-15% больных обнаруживается глубокая олигофрения и ещё у такого же процента больных - «мягкая» умственная отсталость. Большинство пациентов социально адаптированы, выполняют несложную физическую работу. Психологи оценивают их как контактных и доброжелательных. Лишь в тяжёлых случаях таких больных необходимо помещать в специализированные интернаты. Неврологические симптомы включают в себя мышечную гипотонию и в отдельных случаях судороги.
Пороки развития и нарушения других органов сравнительно редки: расщелины нёба, нистагм, страбизм, птоз, катаракта, кривошея в сочетании со сколиозом, кифоз, дефект межпредсердной перегородки.
Помимо определённого комплекса клинических аномалий, синдром Мартина-Белла сопровождается характерной цитогенетической картиной: ломкостью в дистальной части длинного плеча Х-хромосомы (в зоне Xq), что внешне напоминает «спутник» длинного плеча. Эта ломкость выявляется лишь при культивировании лимфоцитов в условиях дефицита фолиевой кислоты, поэтому для обнаружения ломкости нужно либо использовать культуральные среды, лишённые фолиевой кислоты, либо вводить в культуральную среду антагонисты фолиевой кислоты, но даже и при этих условиях ломкость Х-хромосомы выявляется не во всех клетках (до 60%).
Длительное время считалось, что синдром наследуется по Х-сцепленному рецессивному типу. Однако в родословных отмечались случаи тяжело протекающей болезни у женщин и легко протекающей у мужчин. У женщин-гетерозигот отмечали некоторое снижение интеллекта, чаще оцениваемое как пограничная умственная отсталость. У некоторых из них в небольшом проценте клеток обнаруживали ломкую Х-хромосому. Таким образом, наследование синдрома Мартина-Белла не укладывалось в строгие рамки Х-сцепленного рецессивного наследования. Более того, в родословных отмечалась антиципация. Этиология этого заболевания была выяснена с помощью методов молекулярно-генетического анализа. Была обнаружена экспансия нестабильных тринуклеотидных повторов (CGG) в 5'-нетранслируемой области FMR-I гена (fragile menial retardation). В норме в этом гене число повторов варьирует от 6 до 42. Хромосомы, в которых имеется 50-200 повторов, считаются «премутацией». В следующем поколении число повторов может увеличиться (экспансия) до 1000 и более, что и обусловит выраженную клиническую картину, зависящую именно от числа повторов. Соответственно если женщина унаследовала большое число повторов, то она будет больной. Обнаружены ещё два гена, мутации в которых обусловливают такую же клиническую картину, но эти гены встречаются редко.
Диагноз ставят на основании клинической картины и по результатам кли-нико-генеалогического и цитогенетического исследований. Наиболее точный метод - молекулярно-генетическая диагностика.
В настоящее время возможна пренатальная диагностика данного синдрома. Этиотропной терапии пока не существует.
ХРОМОСОМНЫЕ БОЛЕЗНИ
Синдром Дауна
главная / медицинский справочник болезней / детские болезни
 Синдром Дауна – хромосомная аномалия, при которой в кариотипе имеются дополнительные копии генетического материала по 21-ой хромосоме, т. е. наблюдается трисомия по хромосоме 21. Фенотипические признаки синдрома Дауна представлены брахицефалией, плоским лицом и затылком, монголоидным разрезом глазных щелей, эпикантом, кожной складкой на шее, укорочением конечностей, короткопалостью, поперечной ладонной складкой и др. Синдром Дауна у ребенка может быть выявлен пренатально (по данным УЗИ, биопсии ворсин хориона, амниоцентеза, кордоцентеза) или после рождения на основании внешних признаков и генетического исследования. Дети с синдромом Дауна нуждаются в коррекции сопутствующих нарушений развития.
Синдром Дауна – хромосомная аномалия, при которой в кариотипе имеются дополнительные копии генетического материала по 21-ой хромосоме, т. е. наблюдается трисомия по хромосоме 21. Фенотипические признаки синдрома Дауна представлены брахицефалией, плоским лицом и затылком, монголоидным разрезом глазных щелей, эпикантом, кожной складкой на шее, укорочением конечностей, короткопалостью, поперечной ладонной складкой и др. Синдром Дауна у ребенка может быть выявлен пренатально (по данным УЗИ, биопсии ворсин хориона, амниоцентеза, кордоцентеза) или после рождения на основании внешних признаков и генетического исследования. Дети с синдромом Дауна нуждаются в коррекции сопутствующих нарушений развития.
Синдром Дауна
Синдром Дауна - аутосомный синдром, при котором кариотип представлен 47 хромосомами за счет дополнительной копии хромосомы 21-ой пары. Синдром Дауна регистрируется с частотой 1 случай на 500-800 новорожденных. Соотношение полов среди детей с синдромом Дауна составляет 1:1. Впервые синдром Дауна описал английский педиатр Л. Даун в 1866 г., однако хромосомная природа и суть патологии (трисомия по хромосоме 21) была выявлена почти столетие спустя. Клиническая симптоматика синдрома Дауна разнообразна: от врожденных пороков развития и отклонений в умственном развитии до вторичного иммунодефицита. Детям с синдромом Дауна требуется дополнительная медицинская помощь со стороны различных специалистов, в связи с чем они составляют особую категорию в педиатрии.
Причины синдрома Дауна
В норме клетки человеческого организма содержат по 23 пары хромосом (нормальный женский кариотип 46,XX; мужской - 46,XY). При этом одна из хромосом каждой пары наследуется от матери, а другая – от отца. Генетические механизмы развития синдрома Дауна кроются в количественном нарушении аутосом, когда к 21-ой паре хромосом присоединяется дополнительный генетический материал. Наличие трисомии по 21-ой хромосоме определяет черты, характерные для синдрома Дауна.
Появление дополнительной хромосомы может быть обусловлено генетической случайностью (нерасхождением парных хромосом в овогенезе или сперматогенезе), нарушением клеточного деления уже после оплодотворения либо наследованием генетической мутации от матери или отца. С учетом этих механизмов в генетике различают три варианта аномалии кариотипа при синдроме Дауна: регулярную (простую) трисомию, мозаицизм и несбалансированную транслокацию.
Большинство случаев синдрома Дауна (около 94%) связано с простой трисомией (кариотип 47,XX, 21+ или 47,ХY, 21+). При этом три копии 21-ой хромосомы присутствуют во всех клетках вследствие нарушения разделения парных хромосом во время мейоза в материнской или отцовской половых клетках.
Около 1-2% случаев синдрома Дауна приходится на мозаичную форму, которая обусловлена нарушением митоза только в одной клетке зародыша, находящегося на стадии бластулы или гаструлы. При мозаицизме трисомия 21-ой хромосомы выявляется только в дериватах этой клетки, а остальная часть клеток имеет нормальный хромосомный набор.
Транслокационная форма синдрома Дауна встречается у 4-5% пациентов. В этом случае 21-я хромосома либо ее фрагмент прикрепляется (транслоцируется) к какой-либо из аутосом и при мейозе отходит вместе с ней во вновь образовавшуюся клетку. Наиболее частыми «объектами» транслокации служат хромосомы 14 и 15, реже – на 13, 22, 4 и 5. Такая перестройка хромосом может носить случайный характер или наследоваться от одного из родителей, являющегося носителем сбалансированной транслокации и имеющего нормальный фенотип. Если носителем транслокации выступает отец, то вероятность рождения ребенка с синдромом Дауна составляет 3%, если носительство связано с материнским генетическим материалом, риск возрастает до 10-15%.
Факторы риска рождения детей с синдромом Дауна
Рождение ребенка с синдромом Дауна не связано с образом жизни, этнической принадлежностью и регионом проживания родителей. Единственным достоверно установленным фактором, повышающим риск появление ребенка с синдромом Дауна, является возраст матери. Так, если у женщин до 25 лет вероятность рождения больного ребенка составляет 1:1400, к 35 годам уже 1:400, к 40 годам - 1:100; а к 45 - 1:35. Прежде всего, это связано со снижением контроля за процессом деления клеток и увеличением риска нерасхождения хромосом. Однако, поскольку частота родов у молодых женщин в целом выше, то, по статистике, 80% детей с синдромом Дауна рождается от матерей в возрасте до 35 лет. По некоторым данным, возраст отца старше 42-45 лет также увеличивает риск развития синдрома Дауна у ребенка.
Известно, что при наличии синдрома Дауна у одного из однояйцовых близнецов, эта патология в 100% случаев будет иметься у другого. Между тем, у разнояйцовых близнецов, а также братьев и сестер, вероятность такого совпадения ничтожно мала. Среди прочих факторов риска – наличие в роду лиц с синдромом Дауна, возраст матери моложе 18 лет, носительство транслокации одним из супругов, близкородственнные браки, случайные события, нарушающие нормальное развитие половых клеток или зародыша.
Благодаря проведению преимлантационной диагностики, зачатие с помощью ВРТ (в т. ч. экстракорпорального оплдотворения) значительно снижает риск рождения ребенка с синдромом Дауна у родителей из групп риска, однако полностью не исключает такой возможности.
Симптомы синдрома Дауна
Вынашивание плода с синдромом Дауна сопряжено с повышенным риском выкидыша: самопроизвольное прерывание беременности случается примерно у 30% женщин на сроке 6-8 недель. В остальных случаях дети с синдромом Дауна, как правило, рождаются доношенными, но имеют умеренно выраженную гипоплазию (массу тела на 8-10% ниже средней). Несмотря на различные цитогенетические варианты хромосомной аномалии, большинству детей с синдромом Дауна свойственны типичные внешние признаки, позволяющие предположить наличие патологии уже при первом осмотре новорожденного неонатологом. У детей с синдромом Дауна могут быть выражены все или некоторые из физических характеристик, описанных ниже.
80-90% детей с синдромом Дауна имеют черепно-лицевые дизморфии: уплощенное лицо и переносицу, брахицефалию, короткую широкую шею, плоский затылок, деформацию ушных раковин; новорожденные – характерную кожную складку на шее. Лицо отличается монголоидным разрезом глаз, наличием эпикантуса (вертикальной складки кожи, прикрывающей внутренний угол глаза), микрогенией, полуоткрытым ртом часто с толстыми губами и большим высунутым языком (макроглоссией). Мышечный тонус у детей с синдромом Дауна обычно понижен; имеет место гипермобильность суставов (в т. ч. атланто-аксиальная нестабильность),деформация грудной клетки (килевидная или воронкообразная).
Характерными физическими признаками синдрома Дауна служат кроткие конечности, брахидактилия(брахимезофалангия), искривление мизинца (клинодактилия), поперечная («обезьянья») складка на ладони, широкое расстояние между 1 и 2 пальцами стоп (сандалевидная щель) и др. При осмотре детей с синдромом Дауна выявляются белые пятна по краю радужки (пятна Брушфильда), готическое (аркообразное нёбо),неправильный прикус, бороздчатый язык.
При транслокационном варианте синдрома Дауна внешние признаки проявляются отчетливее, чем при простой трисомии. Выраженность фенотипа при мозаицизме определяется долей трисомных клеток в составе кариотипа.
У детей с синдромом Дауна чаще, чем у других в популяции встречаются ВПС (открытый артериальный проток,ДМЖП, ДМПП, тетрада Фалло и др.), косоглазие, катаракта, глаукома, тугоухость, эпилепсия, лейкоз, пороки ЖКТ (атрезия пищевода, стеноз и атрезия двенадцатиперстной кишки, болезнь Гиршпрунга), врожденный вывих бедра. Характерными дерматологическими проблемами пубертатного периода являются сухость кожи, экзема,угревая сыпь, фолликулит.
Дети с синдромом Дауна относятся к часто болеющим; они тяжелее переносят детские инфекции, чаще страдают пневмониями, средними отитами, ОРВИ, аденоидами, тонзиллитом. Слабый иммунитет и врожденные пороки являются наиболее вероятной причиной гибели детей в первые 5 лет жизни.
У большинства больных с синдромом Дауна имеются нарушения интеллектуального развития - как правило, умственная отсталость легкой или средней степени. Моторное развитие детей с синдромом Дауна отстает от сверстников; имеет место системное недоразвитие речи.
Пациенты с синдромом Дауна склонны к развитию ожирения, запоров, гипотиреоза, гнездной алопеции, рака яичка, раннему началу болезни Альцгеймера и др. Мужчины с синдромом Дауна, как правило, бесплодны; фертильность женщин заметно снижена ввиду ановуляторных циклов. Рост взрослых больных обычно на 20 см ниже среднего. Продолжительность жизни составляет около 50-60 лет.
Диагностика синдрома Дауна
Для дородового выявления синдрома Дауна у плода предложена система пренатальной диагностики. Скрининг I триместра проводится на сроке беременности 11-13 недель и включает выявление специфических УЗИ-признаков аномалии и определение уровня биохимических маркеров (ХГЧ, РАРР-А) в крови беременной. Между 15 и 22 неделями беременности выполняется скрининг II триместра: акушерское УЗИ, анализ крови матери на альфа-фетопротеин, ХГЧ и эстриол. С учетом возраста женщины рассчитывается риск рождения ребенка с синдромом Дауна (точность - 56-70%; ложноположительные результаты - 5%).
Беременным из группы риска по рождению ребенка с синдромом Дауна предлагается прохождениепренатальной инвазивной диагностики: биопсии хориона, амниоцентеза или кордоцентеза с кариотипированием плода и консультация медицинского генетика. При получении данных за наличие у ребенка синдрома Дауна решение вопроса о пролонгации или прерывании беременности остается за родителями.
Новорожденные с синдромом Дауна в первые дни жизни нуждаются в провдении ЭхоКГ, УЗИ брюшной полостидля раннего выявления врожденных пороков развития внутренних органов; осмотре детского кардиолога,детского хирурга, детского офтальмолога, детского травматолога-ортопеда.
Медико-педагогическое сопровождение детей с синдромом Дауна
Излечение хромосомной аномалии на сегодняшний день невозможно; любые предлагаемые методы лечения являются экспериментальными и не имеют доказанной клинической эффективности. Однако систематическое медицинское наблюдение и педагогическая помощь детям с синдромом Дауна позволяют добиться успехов в их развитии, социализации и приобретении ими трудовых навыков.
На протяжении всей жизни больные с синдромом Дауна должны находиться под наблюдением специалистов (педиатра, терапевта, кардиолога, эндокринолога, отоларинголога, офтальмолога, невролога и др.) в связи с сопутствующими заболеваниями или повышенным риском их развития. При выявлении тяжелых врожденных пороков сердца и ЖКТ показана их ранняя хирургическая коррекция. В случае выраженного снижения слуха производится подбор слухового аппарата. При патологии органа зрения может потребоваться очковая коррекция,хирургическое лечение катаракты, глаукомы, косоглазия. При гипотиреозе назначается заместительная терапия тиреоидными гормонами и т. д.
Для стимуляции развития моторных навыков показана физиотерапия, занятия ЛФК. Для развития речевых и коммуникативных навыков детям с синдромом Дауна необходимы занятия с логопедом и олигофренопедагогом.
Обучение детей с синдромом Дауна, как правило, осуществляется в специальной коррекционной школе, однако в рамках интегрированного образования такие дети могут посещать и обычную массовую школу. Во всех случаях дети с синдромом Дауна относятся к категории детей с особыми образовательными потребностями, поэтому нуждаются в дополнительной помощи учителей и социальных педагогов, использовании специальных образовательных программ, создании благоприятной и безопасной среды. Важную роль играет психолого-педагогическая поддержка семей, где воспитываются «солнечные дети».
Прогноз и профилактика синдрома Дауна
Возможности обучаемости и социализации лиц с синдромом Дауна различны; они во многом зависят от интеллектуальных способностей детей и от усилий, прилагаемых родителями и педагогами. В большинстве случаев детям с синдромом Дауна удается привить минимальные бытовые и коммуникационные навыки, необходимые в повседневной жизни. Вместе с тем, известны случаи успехов таких пациентов в области изобразительного искусства, актерского мастерства, спорта, а также получения высшего образования. Взрослые с синдромом Дауна могут вести самостоятельную жизнь, овладевать несложными профессиями, создавать семьи.
О профилактике синдрома Дауна можно говорить только с позиции уменьшения возможных рисков, поскольку вероятность рождения больного ребенка существует в любой паре. Акушеры-гинекологи советуют женщинам не откладывать беременность на поздний возраст. Прогнозировать рождение ребенка с синдромом Дауна призвано помочь генетическое консультирование семей и система пренатального скрининга.
Синдром Патау
главная / медицинский справочник болезней / детские болезни
 Синдром Патау – хромосомное заболевание, обусловленное наличием дополнительной копии 13-ой хромосомы (трисомия по 13-ой хромосоме). В структуру синдрома Патау входят множественные дефекты нервной системы (микроцефалия, голопрозэнцефалия), глаз (микрофтальмия, катаракта), костно-мышечной системы (полидактилия, расщелины губы и нёба, омфалоцеле), сердца, урогенитальной системы и др. Для выявления и подтверждения синдрома Патау проводится пренатальный скрининг, исследование кариотипа ребенка после рождения. Детям с синдромом Патау необходима общеукрепляющая терапия; по показаниям – коррекция врожденных пороков развития.
Синдром Патау – хромосомное заболевание, обусловленное наличием дополнительной копии 13-ой хромосомы (трисомия по 13-ой хромосоме). В структуру синдрома Патау входят множественные дефекты нервной системы (микроцефалия, голопрозэнцефалия), глаз (микрофтальмия, катаракта), костно-мышечной системы (полидактилия, расщелины губы и нёба, омфалоцеле), сердца, урогенитальной системы и др. Для выявления и подтверждения синдрома Патау проводится пренатальный скрининг, исследование кариотипа ребенка после рождения. Детям с синдромом Патау необходима общеукрепляющая терапия; по показаниям – коррекция врожденных пороков развития.
Синдром Патау
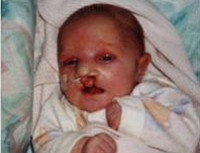 Синдром Патау - хромосомная аномалия, представляющая собой трисомию по 13-ой паре аутосом. Синдрома Патау также встречается в литературе под названиями трисомия D и трисомия 13. Частота рождения детей с синдромом Патау составляет 1:7000-10000; соотношение полов примерно одинаковое. Клинический симптомокомплекс был описан еще в XVII веке; связь же заболевания с увеличением количества хромосом 13-ой пары была установлена в 1960 г. К. Патау, по имени которого данный синдром и получил свое название. При синдроме Патау у ребенка имеются множественные и крайне тяжелые аномалии развития, определяющие частые случаи внутриутробной гибели плода и малую продолжительность жизни детей с данной патологией.
Синдром Патау - хромосомная аномалия, представляющая собой трисомию по 13-ой паре аутосом. Синдрома Патау также встречается в литературе под названиями трисомия D и трисомия 13. Частота рождения детей с синдромом Патау составляет 1:7000-10000; соотношение полов примерно одинаковое. Клинический симптомокомплекс был описан еще в XVII веке; связь же заболевания с увеличением количества хромосом 13-ой пары была установлена в 1960 г. К. Патау, по имени которого данный синдром и получил свое название. При синдроме Патау у ребенка имеются множественные и крайне тяжелые аномалии развития, определяющие частые случаи внутриутробной гибели плода и малую продолжительность жизни детей с данной патологией.
Причины синдрома Патау
Основой для развития синдрома Патау служит присутствие в кариотипе дополнительной копии 13-ой хромосомы. В большинстве случаев (75-80%) имеет место простая полная трисомия, связанная с нерасхождением 13-ой хромосомы в мейозе у одного из родителей (чаще у матери). При этом все без исключения клетки плода имеют кариотип 47,XX 13+ или 47,XY 13+. Меньшая часть случаев синдрома Патау представлена несбалансированными транслокациями хромосом 13-й пары, мозаичными формами, изохромосомой.
Точные причины утроения 13-ой хромосомы не установлены. Известно лишь, что генетический сбой может произойти во время формирования гамет или уже на этапе образования зиготы. Прослеживается связь между частотой развития синдрома Патау у плода и возрастом матери, хотя эта зависимость менее выражена, чем присиндроме Дауна. Роль других факторов (инфекций, соматических заболеваний матери, вредных привычек, экологического неблагополучия и пр.) достоверно не определена.
Генетическая мутация в гаметогенезе или зародышевой клетке в основном возникает de novo, как случайное событие. Наследственные формы синдрома Патау связаны с наличием робертсоновской (сбалансированной) транслокации у родителей. Вновь возникшая робертсоновская транслокация может наследоваться, не вызывая синдром Патау у ребенка, но увеличивая риск рождения детей с данной аномалией в последующих поколениях.
Симптомы синдрома Патау
Синдром Патау сопровождается формированием множественных тяжелых пороков, нередко приводящих к внутриутробной гибели плода. Почти в половине случаев беременность плодом с синдромом Патау осложняется многоводием.
Дети обычно рождаются в срок, но с маленьким по отношению к сроку гестации весом - около 2500 г (т. н. пренатальной гипотрофией). Роды нередко осложняются асфиксией новорожденного. У ребенка с синдромом Патау выявляются врожденные аномалии разви






