Вирильная форма обусловлена дефицитом 21-гидроксилазы. При этом происходит нарушение синтеза только глюкокортикоидов, компенсация вирильной формы происходит благодаря гиперплазии надпочечников, в результате возникает латентная надпочечниковая недостаточность. Гиперпродукция андрогенов, возникающая в период внутриутробного развития ребенка, ведет к андрогенизации половых признаков (вторичных) плода, вследствие чего рождаются девочки, у которых обнаруживаются признаки ложного женского гермафродитизма (мошонкообразные большие половые губы, пенисообразный клитор), однако изменения внутренних половых органов под влиянием андрогенов не происходит: яичники и матка, как правило, развиваются в соответственно возрастной норме. Мальчики рождаются с увеличенным половым членом. Кроме того, появляется гиперпигментация наружных половых органов, ареол вокруг сосков, кожных складок, анального отверстия.
Заболевание также проявляется макросомией, которая возникает из-за анаболического действия избытка андрогенов и проявляется увеличением массы тела и роста новорождённых. Ее можно наблюдать у представителей обоих полов. Больные дети с первых лет жизни растут быстрее сверстников. Однако, начиная с 12—14 лет рост костей (трубчатых) заканчивается, поэтому дети остаются низкорослыми, телосложение непропорционально, сильно развита мускулатура (ребенок-«Геркулес»).
Ранним проявлением адреногенитального синдрома является гирсутизм — избыточный рост волос на теле ребенка по мужскому типу (появляется в возрасте от двух до пяти лет), имеет вид чрезмерного оволосения лица (борода, усы), лобовой области, подмышечных впадина, груди, спины, конечностей. Данное проявление возникает в результате гиперпродукции андрогенов с реализацией их эффектов.
Еще одним признаком данной патологии является маскулинизация, которая возникает в результате действия избыточного количества андрогенов, циркулирующих в крови, на ткани-мишени и клетки – мишени. Вследствие этого мужские вторичные половые признаки развиваются у особи генетически женского пола. Проявляется маскулинизация гипотрофией или полной атрофией матки и молочных желез, нарушениями менструального цикла или полным отсутствием менструаций (аменорея), телосложением мужского типа, изменением поведения (стремления к лидерству, появление властолюбия, увлечение техникой, мужскими хобби и так далее), низким тембром голоса.
Для сольтеряющей формы характерна стойкое снижение АД (артериальная гипотензия) вплоть до развития коллапса. Причинами ее возникновения являются гиповолемия, гипогидратация, гипонатриемия, гиперкалиемия – все это возникает вследствие дефицита альдостерона, из-за чего происходит нарушение водно-обменных функций.
Для гипертензивной формы характерностойкое увеличение АД(артериальная гипертензия). Оно обусловлено избытком в крови минералокортикоидов.
Важно отметить, что зачастую люди с таким диагнозом бесплодны.
Диагностика
§ Сбор анамнестических данных
§ Клиническое обследование
§ Рентгенологическое исследование
§ УЗИ
§ Определение полового хроматина
§ Анализ мочи на 17-кетостероиды (повышенное их выделение с мочой)
§ Осмотр эндокринолога
§ Осмотр гинеколога
§ Осмотр уролога
§ Осмотр онколога и врачей других специальностей при необходимости
Лечение
Лечение адреногенитального синдрома включает в себя пожизненную заместительную терапию глюкокортикоидами. Доза препаратов подбирается индивидуально под контролем в суточной моче 17-кетостероидов. При сольтеряющей форме в дополнение назначают минералокортикоиды, а также хлорид натрия. При гипертонической форме в рационе ограничивают хлорид натрия (не более 3 грамм в сутки). При желании родителей или больного проводится пластика наружных половых органов – клиторэктомия, пластика влагалища. Огромное значение в лечении адреногенитального синдрома имеет психотерапия. Если начать лечение своевременно – для жизни прогноз благоприятный.
Болезнь Вильсона
главная / медицинский справочник болезней / нервные болезни

Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона может протекать в брюшной, ригидно-аритмогиперкинетической, дрожательной или экстрапирамидно-корковой форме. Диагностика болезни Вильсона включает офтальмологическое обследование, биохимические анализы мочи и крови, МРТ или КТ головного мозга. Основу патогенетической терапии составляют тиоловые препараты, которые могут приниматься в течении нескольких лет и даже пожизненно.
Болезнь Вильсона

Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФ-азы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях.
Первооткрыватель заболевания — А.К. Вильсон, описавший заболевание в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году. Понятию «болезнь Вильсона» соответствуют также: болезнь Вильсона-Коновалова, болезнь Вестфаля-Вильсона-Коновалова, дистрофия гепатоцеребральная, дистрофия гепатолентикулярная, дегенерация лентикулярная прогрессирующая.
Классификация болезни Вильсона
Согласно классификации Н.В. Коновалова различают пять форм болезни Вильсона:
· брюшная
· ригидно-аритмогиперкинетическая
· дрожательно-ригидная
· дрожательная
· экстрапирамидно-корковая
Этиология и патогенез болезни Вильсона
Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3-q21.1). Организм человека содержит около 50-100 мг меди. Суточная потребность меди для человека — 1-2 мг. 95% абсорбированной в кишечнике меди, транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5% в форме комплекса с альбумином. Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохром-С-оксидаза и др.). При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтез главного медьсвязывающего белка (церулоплазмина) и выведение меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению.
Клиническая картина болезни Вильсона
Для болезни Вильсона характерен клинический полиморфизм. Первые проявления заболевания могут появиться в детстве, юношестве, в зрелом возрасте и гораздо реже в зрелом возрасте. В 40-50% случаев Болезнь Вильсона манифестирует с поражения печени, в остальных — с психических и неврологических расстройств. С вовлечением в патологический процесс нервной системы обнаруживается кольцо Кайзера-Флейшера.
Брюшная форма болезни Вильсона развивается преимущественно до 40 лет. Характерный признак — тяжелое поражение печени по типу цирроза печени, хронического гепатита, фульминантного гепатита.
Ригидно-аритмогиперкинетическая форма болезни Вильсона манифестирует в детском возрасте. Начальные проявления — мышечная ригидность, амимия, смазанность речи, трудности при выполнении мелких движений, умеренное снижение интеллекта. Для этой формы заболевания характерно прогрессирующее течение, с наличием эпизодов обострения и ремиссии.
Дрожательная форма болезни Вильсона возникает в возрасте от 10 до 30 лет. Преобладающим симптомом является тремор. Кроме того, могут наблюдаться брадикинезия, брадилалия, тяжелый психоорганический синдром, эпилептические приступы.
Экстрапирамидно-корковая форма болезни Вильсона встречается весьма редко. Ее начало схоже с началом какой-либо из вышеперечисленных форм. Для нее характерны эпилептические припадки, экстрапирамидные и пирамидные нарушения и выраженный интеллектуальный дефицит.
Диагностика болезни Вильсона
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера.Биохимические исследования обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
Дифференциальный диагноз
При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца. Основным дифференциально-диагностическим признаком этих заболеваний является отсутствие характерных для болезни Вильсона кольца Кайзера-Флейшера и расстройств обмена меди.
Лечение болезни Вильсона
Основой патогенетического лечения болезни Вильсона является назначение тиоловых препаратов, в первую очередь — D-пеницилламина (купренил) либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза). Лечение купренилом проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол (5 ml в/м) назначают в случае непереносимости (плохой переносимости) купренила. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — синемент, циклодол.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень).
Немедикаментозное лечение болезни Вильсона состоит в назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.).
Прогноз и профилактика болезни Вильсона
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме. Постоянный прием тиоловых препаратов по схеме, назначенной врачом-специалистом, позволяет поддерживать профессиональную и социальную активность пациента.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.
X-сцепленные рецессивные
Гемофилия 
Гемофилия – наследственная патология системы гемостаза, в основе которой лежит снижение или нарушение синтеза VIII, IX или XI факторов свертывания крови. Специфическим проявлением гемофилии служит склонность больного к различным кровотечениям: гемартрозам, внутримышечным и забрюшинным гематомам, гематурии, желудочно-кишечным кровотечениям, длительным кровотечениям при операциях и травмах и др. В диагностике гемофилии первостепенное значение имеет генетическое консультирование, определение уровня активности факторов свертываемости, ДНК-исследование, анализ коагулограммы. Лечение гемофилии предполагает проведение заместительной терапии: трансфузии гемоконцентратов с факторами свертывания VIII или IX, свежезамороженной плазмы, антигемофильного глобулина и др.
Гемофилия
Гемофилия – заболевание из группы наследственных коагулопатий, обусловленное дефицитом факторов свертывания плазмы крови и характеризующееся повышенной склонностью к геморрагиям. Распространенность гемофилии А и В составляет 1 случай на 10000-50000 представителей мужского пола. Чаще всего дебют заболевания приходится на ранний детский возраст, поэтому гемофилия у ребенка является актуальной проблемой педиатрии и детской гематологии. Кроме гемофилии, у детей встречаются и другие наследственные геморрагические диатезы: геморрагическая телеангиэктазия, тромбоцитопатия, болезнь Гланцмана т. п.
Причины гемофилии
Гены, обусловливающие развитие гемофилии, сцеплены с половой Х-хромосомой, поэтому заболевание наследуется по рецессивному признаку по женской линии. Наследственной гемофилией болеют практически исключительно лица мужского пола. Женщины являются проводниками (кондукторами, носителями) гена гемофилии, передающими заболевание части своих сыновей.
У здорового мужчины и женщины-кондуктора с одинаковой вероятностью могут родиться как больные, так и здоровые сыновья. От брака мужчины, больного гемофилией со здоровой женщиной рождаются здоровые сыновья или дочери-кондукторы. Описаны единичные случаи гемофилии у девочек, рожденных от матери-носителя и больного гемофилией отца.
Врожденная гемофилия встречается почти у 70 % пациентов. В этом случае наследуется форма и тяжесть гемофилии. Около 30% наблюдений приходится на спорадические формы гемофилии, связанные с мутацией в локусе, кодирующем синтез плазменных факторов свертывания крови на Х-хромосоме. В дальнейшем такая спонтанная форма гемофилии становится наследственной.
Свертываемость крови, или гемостаз, служит важнейшей защитной реакцией организма. Активизация системы гемостаза происходит в случае повреждения сосудов и начала кровотечения. Свертываемость крови обеспечивается тромбоцитами и особыми веществами – плазменными факторами. При дефиците того или иного фактора свертывания своевременный и адекватный гемостаз становится невозможным. При гемофилии в связи с дефицитом VIII, IX или других факторов нарушается первая фаза свертывания крови - образование тромбопластина. При этом увеличивается время свертывания крови; иногда кровотечение не останавливается в течение нескольких часов.
Классификация гемофилии
В зависимости от дефицита того или иного фактора свертываемости крови, различают гемофилию А (классическую), В (болезнь Кристмаса), С и др.
Классическая гемофилия составляет подавляющее большинство (около 85%) случаев синдрома и связана с дефицитом VIII фактора свертывания (антигемофильного глобулина), приводящим к нарушению образование активной тромбокиназы.
При гемофилии В, составляющей 13% случаев заболевания, имеет место недостаток IX фактора (плазменного компонента тромбопластина, фактора Кристмаса), также участвующего в образовании активной тромбокиназы в I фазе свертывания крови.
Гемофилия С встречается с частотой 1-2% и обусловлена недостаточностью XI фактора свертывания крови (предшественника тромбопластина). На остальные разновидности гемофилии приходится менее 0,5% случаев; при этом может отмечаться дефицит различных плазменных факторов: V (парагемофилия), VII (гипопроконвертинемия), Х (болезнь Стюарта – Прауэр) и др.
Тяжесть клинического течения гемофилии зависит от степени недостаточности коагуляционной активности плазменных факторов свертывания крови.
При гемофилии тяжелой степени уровень недостающего фактора составляет до 1%, что сопровождается развитием тяжелого геморрагического синдрома уже в раннем детском возрасте. У ребенка с тяжелой гемофилией возникают частые спонтанные и посттравматические кровоизлияния в мышцы, суставы, внутренние органы. Сразу после рождения ребенка могут обнаруживаться кефалогематомы, длительные кровотечения из пуповинного отростка, мелена; позднее - продолжительные кровотечения, связанные с прорезыванием и сменой молочных зубов.
При среднетяжелой степени гемофилии у ребенка уровень плазменного фактора составляет 1-5%. Заболевание развивается в дошкольном возрасте; геморрагический синдром выражен умеренно, отмечаются кровоизлияния в мышцы и суставы, гематурия. Обострения случаются 2-3 раза в год.
Легкая форма гемофилии характеризуется уровнем фактора выше 5%. Дебют заболевания возникает в школьном возрасте, часто в связи с травмами или операциями. Кровотечения более редкие и менее интенсивные.
Симптомы гемофилии
У новорожденных детей признаками гемофилии могут служить длительное кровотечение из культи пуповины, подкожные гематомы, кефалогематомы. Кровотечения у детей первого года жизни могут быть связаны спрорезыванием зубов, оперативными вмешательствами (инцизией уздечки языка, циркумцизио). Острые края молочных зубов могут стать причиной прикусывания языка, губ, щек и кровотечений из слизистых оболочек полости рта. Однако в грудном возрасте гемофилия дебютирует редко в связи с тем, что материнском молоке содержится достаточное количество активной тромбокиназы.
Вероятность посттравматических кровотечений значительно возрастает, когда ребенок с гемофилией начинает вставать и ходить. Для детей после года характерны носовые кровотечения, подкожные и межмышечные гематомы, кровоизлияния в крупные суставы. Обострения геморрагического диатеза случаются после перенесенных инфекций (ОРВИ, ветрянки, краснухи, кори, гриппа и др.) вследствие нарушения проницаемости сосудов. В этом случае нередко возникают самопроизвольные диапедезные геморрагии. Ввиду постоянных и длительных кровотечений у детей с гемофилией имеется анемия различной степени выраженности.
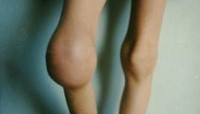
По степени убывания частоты кровоизлияния при гемофилии распределяются следующим образом: гемартрозы(70—80%), гематомы (10-20%), гематурия (14-20%), желудочно-кишечные кровотечения (8%), кровоизлияния в ЦНС (5%).
Гемартрозы являются наиболее частым и специфическим проявлением гемофилии. Первые внутрисуставные кровоизлияния у детей с гемофилией случаются в возрасте 1-8 лет после ушибов, травм или спонтанно. При гемартрозе выражен болевой синдром, отмечается увеличение сустава в объеме, гиперемия и гипертермия кожи над ним. Рецидивирующие гемартрозы приводят к развитию хронического синовита, деформирующего остеоартроза и контрактур. Деформирующий остеоартроз приводит к нарушению динамики опорно-двигательного аппарата в целом (искривлению позвоночника и таза, гипотрофии мышц, остеопорозу, вальгусной деформации стопы и др.) и к наступлению инвалидности уже в детском возрасте.
При гемофилии часто возникают кровоизлияния в мягкие ткани – подкожную клетчатку и мышцы. У детей обнаруживаются непроходящие синяки на туловище и конечностях, часто возникают глубокие межмышечные гематомы. Такие гематомы склонны к распространению, поскольку излившаяся кровь не сворачивается и, проникая вдоль фасций, инфильтрирует ткани. Обширные и напряженные гематомы могут сдавливать крупные артерии и периферические нервные стволы, вызывая интенсивные боли, паралич, атрофию мышц или гангрену.
Довольно часто при гемофилии возникают кровотечения из десен, носа, почек, органов ЖКТ. Кровотечение может быть инициировано любыми медицинскими манипуляциями (внутримышечной инъекцией, экстракцией зуба,тонзиллэктомией и др.). Крайне опасными для ребенка с гемофилией являются кровотечения из зева и носоглотки, поскольку могут привести к обструкции дыхательных путей и потребовать экстренной трахеостомии. Кровоизлияния в мозговые оболочки и головной мозг приводят к тяжелым поражениям ЦНС или летальному исходу.
Гематурия при гемофилии может возникать самопроизвольно или вследствие травм поясничной области. При этом отмечаются дизурические явления, при образовании кровяных сгустков в мочевыводящих путях - приступыпочечной колики. У больных с гемофилией нередко обнаруживаются пиелоэктазия, гидронефроз, пиелонефрит.
Желудочно-кишечные кровотечения у пациентов с гемофилией могут быть связаны с приемом НПВС и др. лекарств, с обострением латентного течения язвенной болезни желудка и двенадцатиперстной кишки,эрозивным гастритом, геморроем. При кровоизлияниях в брыжейку и сальник развивается картина острого живота, требующая дифференциальной диагностики с острым аппендицитом, кишечной непроходимостью и др.
Характерным признаком гемофилии является отсроченный характер кровотечения, которое обычно развивается не сразу после травмы, а через некоторое время, иногда спустя 6-12 и более часов.
Диагностика гемофилии
Диагностика гемофилии проводится при участии ряда специалистов: неонатолога, педиатра, генетика, гематолога. При наличии у ребенка сопутствующей патологии или осложнений основного заболевания проводятся консультации детского гастроэнтеролога, детского травматолога-ортопеда, детского отоларинголога,детского невролога и др.
Супружеские пары, находящиеся в группе риска по рождению ребенка с гемофилией, должны пройти медико-генетическое консультирование еще на этапе планирования беременности. Выявить носительство дефектного гена позволяет анализ генеалогических данных и молекулярно-генетическое исследование. Возможно проведение пренатальной диагностики гемофилии с помощью биопсии хориона или амниоцентеза и исследования ДНК клеточного материала.
После рождения ребенка диагноз гемофилии подтверждается с помощью лабораторных исследований гемостаза. Основные изменения показателей коагулограммы при гемофилии представлены увеличением времени свертывания крови, АЧТВ, тромбинового времени, МНО, времени рекальцификации; уменьшением ПТИ и др. Решающее значение при диагностике формы гемофилии принадлежит определению снижения прокоагулянтной активности одного из факторов свертывания ниже 50%.
При гемартрозах ребенку с гемофилией проводится рентгенография суставов; при внутренних кровотечениях и забрюшинных гематомах – УЗИ брюшной полости и забрюшинного пространства; при гематурии – общий анализ мочи и УЗИ почек и т. д.
Лечение гемофилии
При гемофилии полное избавление от заболевания невозможно, поэтому основу лечения составляет заместительная гемостатическая терапия концентратами VIII и IX факторов свертывания крови. Необходимая доза концентрата определяется степенью выраженности гемофилии, тяжестью и видом кровотечения.
В лечении гемофилии выделяют два направления – профилактические и «по требованию», в период проявлений геморрагического синдрома. Профилактическое введение концентратов факторов свертывания крови показано пациентам с тяжелой формой гемофилии и проводится 2-3 раза в неделю для предупреждения развития гемофилической артропатии и прочих кровотечений. При развитии геморрагического синдрома требуются повторные трансфузии препарата. Дополнительно используются свежезамороженная плазма, эритромасса, гемостатики. Все инвазивные вмешательства у больных с гемофилией (наложение швов, удаление зубов, любые операции) проводятся под прикрытием гемостатической терапии.
При незначительных наружных кровотечениях (порезах, кровотечениях из полости носа и рта) может использоваться гемостатическая губка, наложение давящей повязки, обработка раны тромбином. При неосложненном кровоизлиянии ребенку необходим полный покой, холод, иммобилизация больного суставагипсовой лонгетой, в дальнейшем – УВЧ, электрофорез, ЛФК, легкий массаж. Больным с гемофилией рекомендуется диета, обогащенная витаминами А, В, С, D, солями кальция и фосфора.
Прогноз и профилактика гемофилии
Длительная заместительная терапия приводит к изоиммунизации, образованию антител, блокирующих прокоагулянтную активность вводимых факторов, и неэффективности гемостатической терапии в обычных дозах. В таких случаях больному с гемофилией проводится плазмаферез, назначаются иммунодепрессанты. Поскольку больным с гемофилией проводится частое переливание компонентов крови, не исключается риск инфицирования ВИЧ-инфекцией, гепатитами В, С и D, герпесом, цитомегалией.
Легкая степень гемофилии не влияет на продолжительность жизни; при тяжелой гемофилии прогноз ухудшается при массивных кровотечениях, обусловленных операциями, травмами.
Профилактика предполагает проведение медико-генетического консультирования супружеских пар, имеющих отягощенный семейный анамнез по гемофилии. Дети, больные гемофилией, всегда должны иметь при себе специальный паспорт, где указан тип заболевания, группа крови и Rh-принадлежность. Им показан охранительный режим, профилактика травм; диспансерное наблюдение педиатра, гематолога, детского стоматолога, детского ортопеда и др. специалистов; наблюдение в условиях специализированного гемофильного центра.
Мышечная дистрофия Дюшенна/Беккера.
Тип наследования.
Х-сцепленный рецессивный, т.е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена дистрофина, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Приблизительно 30% всех случаев заболевания связаны с возникновением свежих мутаций в гене дистрофина, а остальные 70% обусловлены носительством матерью пробанда патологической мутации в одной из Х хромосом. Считается, что 6-7% всех спорадических случаев заболевания являются следствием гонадного мозаицизма - существования в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллелями гена дистрофина.






