

Индивидуальные очистные сооружения: К классу индивидуальных очистных сооружений относят сооружения, пропускная способность которых...

Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Индивидуальные очистные сооружения: К классу индивидуальных очистных сооружений относят сооружения, пропускная способность которых...
Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Топ:
Характеристика АТП и сварочно-жестяницкого участка: Транспорт в настоящее время является одной из важнейших отраслей народного хозяйства...
Характеристика АТП и сварочно-жестяницкого участка: Транспорт в настоящее время является одной из важнейших отраслей народного...
Особенности труда и отдыха в условиях низких температур: К работам при низких температурах на открытом воздухе и в не отапливаемых помещениях допускаются лица не моложе 18 лет, прошедшие...
Интересное:
Искусственное повышение поверхности территории: Варианты искусственного повышения поверхности территории необходимо выбирать на основе анализа следующих характеристик защищаемой территории...
Принципы управления денежными потоками: одним из методов контроля за состоянием денежной наличности является...
Подходы к решению темы фильма: Существует три основных типа исторического фильма, имеющих между собой много общего...
Дисциплины:
|
из
5.00
|
Заказать работу |
|
|
|
|
Абсолютная и относительная погрешности.
Абсолютная погрешность – отклонение результата измерения от истинного значения измеряемой величины: ΔXͥi=Xi-µ, где Xi – измеряемая величина, µ - истинное значение измеряемой или анализируемой величины.
В большинстве случаев µ неисвестно, и абсолютная погрешность не может быть точно определена, поэтому часто определяют верхнюю границу ΔXͥi.
Предельная абсолютная погрешность ΔX приближенно ΔXͥi является наименьшим числом: ΔX≥l ΔXͥil, тогда интервал, в котором заключено точное значение числа µ будет находится в этих пределах:
Х- ΔX≤µ≤ Х+ΔX, где Х – измеряемая величина.
Абсолютные погрешности не полностью характеризуют отклонение величины от истинного значения.
ΔX=1, но в первом случае µ1=10, а во втором µ2=1000.
Поэтому используют понятие относительной погрешности (ошибки), кот представ собой отношение абсолют погрешности к истинному значению измеряемой величины: Ɛ= ΔX/µ.
Обычно относительная погрешность измер в %: Ɛ= ΔX/µ*100%.
2 Классификация погрешностей по форме представления:
- абсолютна я - отклонение результата измерения от истинного назначения измеряемой величины. ∆Xi=Xi-µ
- относительная - отношение абсолютной погрешности к истинному значению. E=∆X/µ
- приведённая - погрешность, выраженная отношением абсолютной погрешности средства измерений к условно принятому значению величины, постоянному во всем диапазоне измерений или в части диапазона. E =∆X/ µn, где µn - нормирующее значение, которое зависит от типа шкалы измерительного прибора и определяется по его градуировке.
Погрешности измерений и анализа по характеру проявлений условно подразделяют на 3 группы: Систематические Случайные Грубые
|
|
Систематические – погрешности, которая при повторных измерениях остается постоянной или закономерно изменяется. К ним можно отнести приборные погрешности, обусловленные несовершенством конструкции прибора.
Систематические погрешности по причине возникновения делятся на инструментальные, методические, оперативные и личностные погрешности и погрешности предубеждения.
Инструментальные: источник погрешности – некалиброванные приборы и непроверенные реактивы.
Методические: погрешности метода зависят от св-в анализируемой системы. Методические погрешности дают расчетные формулы, кот использ для вычисления величин на основании замеренных данных. Такие погрешности часто остаются не обнаружены.
Оперативные и личностные погрешности: они связаны с операциями, выполняемыми в ходе анализа и зависят от квалификации исследователя и его способностей.
Погрешности предубеждения: они заключаются в том, что при повторных определениях исследователь из 2-ух равновероятных показателей прибора при отсчете на глаз выбирает то значение, кот находится ближе к предыдущему результату.
Случайная погрешность – погрешность, кот при повторном измерении изменяется случайным образом. Они обнаруживаются в нерегулярных расхождениях результатов измерений, обычно в последних двух-трех цифрах. Знак такой погрешности может меняться от опыта к опыту. Причины случайных погрешностей многообразны: шумы измерительного прибора, вариация его показаний, случайные колебания параметров электрической сети и условий измерений, погрешности округления отсчетов и многие другие.
Грубые ошибки – погрешности, которые существенно превышают ожидаемые погрешности при данных условиях исследования. Такие грубые ошибки часто бывают следствием грубых оперативных погрешностей аналитика. Они также возникают при использ неисправной измерительной аппаратуры, несоблюдения условий измерения, Возможной причиной промаха могут быть сбои работе технических средств, а также кратковременные резкие изменения условий измерений. Результаты измерений, содержащих промах, выявляются и во внимание не принимаются.
|
|
Выполнение анализа независимыми методами.
Анализ проводят двумя или несколькими различными независимыми методами. Если результаты одинаковы, то м.б.уверенным, что анализ выполнен правильно, а в дальнейшем с достаточной точностью.
Метод стандартных образцов
Стандартный образец – различный материал, содержание определяемых элементов в котором известно с высокой точностью. Каждый станд.образец снабжается определённым документом (паспортом). Чтобы установить правильность анализа какого-либо материала из имеющего набора стандартных образцов, выбирают тот, который по-своему составу наиболее близок к пробе.
Случайные погрешности
Случайной погрешностью измерения называется составляющая погрешности измерения, изменяющаяся случайным образом (по знаку и значению) при повторных измерениях одной и той же величины. Наличие случайных погрешностей выявляется при проведении ряда измерений постоянной физической величины, когда оказывается, что результаты измерений не совпадают друг с другом. Причины случайных погрешностей многообразны: шумы измерительного прибора, вариация его показаний, случайные колебания параметров электрической сети и условий измерений, погрешности округления отсчетов и многие другие.
Название случайной погрешности указывает на отсутствие известной причины, закономерностей и появлений. Мы можем лишь только предполагать о наличии причины. Мероприятия направленные на устранение этих эффектов и явлений во многих случаях позволяют уменьшить случ.погрешность, но не устранить полностью. Появление случайной погрешности рассматривается как внезапность, поэтому эти погрешности подвергаются обработке. Правильность измерений – качество измерений, отражающее близость к нулю систематич. погрешностей. Сходимость измерений – качество измерений, отражающее близость друг к другу результатов измерений, выполняемых в одинаковых условиях. Воспроизводимость – качество измерений, отражающ. близость друг к другу результатов измерений, выполняемых в различных условиях (в разное время и место). Точность измерений – качество измерений, отражающ. близость их результатов к истинному значению измеряемой величины. Высокая точность измерений соответствует малым погрешностям всех видов, как систематических, так и случайных. Количественно точность может быть выражена обратной величиной модуля относит. погрешности. Напр, если относит. погрешность = 0.0002, то точность = 1/0.0002=5000. Значение физич. величины, найденное экспериментально и настолько приближающееся к истинному значению, что для данной цели может быть использовано вместо него, называют действительным значением величины.
|
|
Нормальное распределение
Свойство большого числа малых случайных погрешностей описываются нормальным распределением (уравнением Гаусса). Говорят, что случайная величина нормально распределена или подчиняется закону распределения Гаусса, если ее плотность распределения  имеет вид
имеет вид

где a - любое действительное число, а  >0.
>0.
Физическая величина, подверженная влиянию значительного числа независимых факторов, способных вносить с равной погрешностью положительные и отрицательные отклонения, вне зависимости от природы этих случайных факторов, часто подчиняется нормальному распределению, поэтому из всех распределений в природе чаще всего встречается нормальное (отсюда и произошло одно из названий этого распределения вероятностей).

Свойства:
1вероятность попадания СВ Х в интервал от (-∞;+)=1, следует, что площадь ограничена прямой и осью Х=1.
2.если через 2 значения Х:Х1 и Х2 провести вертикали до пересечения с прямой, то площадь, ограниченная Х1 и Х2 и кривой называется доверительной вероятностью.
Распределение Стьюдента
t-распределение Стьюдента - это непрерывное одномерное распределение с одним параметром - количеством степеней свободы.
Распределение Стьюдента по сути представляет собой сумму нескольких нормально распределенных случайных величин. Чем больше величин, тем больше верятность, что их сумма будет иметь нормальное распределение. (t-распределение) Таким образом, количество суммруемых величин определяет важнейший параметр формы данного распредения - число степеней свободы. Распределение Стьюдента сходится к стандартному нормальному при  .
.
|
|
 Плотность распределения Стьюдента по сравнению с плотностью стандартного нормального распределения.
Плотность распределения Стьюдента по сравнению с плотностью стандартного нормального распределения.
Согласно данному распределению погрешность среднеарифметическая или относительное стандартное отклонение рассчитывается: S=t p,f*Sx/корень n, где t p,f – коэф.стьюдента, который зависит от f и p, p-доверительная вероятность. Sx/корень n – стандартное отклонение среднего значения.
Коэф.находят в специальных статистических таблицах. Из этих таблиц можно увидеть, что наиболее эффективно сужение доверит интервала наблюдается при увеличении числа значений n до 4-5. Если задана доверит.вер-ть р, то доверит.интервал можно легко определить и тогда искомая величина будет записана <x>±S. Обычно доверит.интервал анализа вычисляют при доверит.вер-ти p=0,95.
Требование к пробе анализа. Генеральная, средняя, лабораторная и анализируемая проба
Все вещества, которые поступают на предприятия для их дальнейшей переработки образуют генеральную или первичную совокупность. Поскольку поступает очень большое количество, то проводят отбор небольшой части этих веществ, т.е.отбор проб. В этом случае проба является частью всей генеральной сов-ти.Основное требование к пробе материала, поступающей в лабораторию - ее представительность, т.е.состав и свойства пробы и всей генеральной сов-ти должны бать близкими. Требование к пробам, находящимся в стадии анализа, – не допустить загрязнения в процессе пробоподготовки. В процессе отбора и хранения пробы возможны потери определяемых компонентов (особенно летучих компонентов), внесение загрязнений, изменения химического состава. Поэтому следует строго регламентировать методику пробоотбора: число и последовательность операций измельчения и просеивания, температурный режим, время растирания и контакта с атмосферой, материал пробоотборников и измельчающих устройств, способы очистки и т.д. Пробы, поступающие в лабораторию, осматривают, вскрывают упаковку и регистрируют в журнале в соответствии с сопроводительной документацией, удостоверяющей объект.
Первичную или генеральную пробу отбирают на первом этапе от большой массы материала, лабораторную или паспортную пробу получают после уменьшения массы генеральной пробы до массы, необходимой для проведения всего анализа полностью, аналитическую пробу отбирают от лабораторной для проведения единичного определения. Средней пробой называют относительно небольшое количество исследуемого материала, которое по своему составу, физическим и физико-химическим свойствам соответствует всей партии.Перед отбором генеральной пробы необходимо определить ее представительность, а при получении лабораторной, кроме того, рассчитать массу пробы, позволяющую провести весь анализ.
|
|
Метод разложения проб
Под минерализацией в химическом анализе понимается разложение органических веществ и материалов на их основе с целью выделения определяемых элементов в виде устойчивых неорганических соединений, удобных для последующего анализа.
Среди обычных методов разрушения органических компонент следует выделить способ сухой минерализации, способ мокрой минерализации, способ кислотной экстракции.
Способ сухой минерализации основан на полном разложении органических веществ путём сжигания пробы сырья или продуктов в электропечи при контролируемом температурном режиме и предназначен для всех видов сырья и продовольственных продуктов, кроме животных, растительных жиров и масел.
Способ мокрой минерализации основан на полном разложении органических вещ-в пробы продукта при нагревании с серной и азотной концентрированными кислотами с добавлением хлорной кислоты или перекиси водорода и предназнач.для всех видов сырья и продуктов, кроме сливочного масла и животных жиров.
Способ кислотной экстракции основан на экстракции токсичных элементов из пробы продукта кипячением с разбавлением соляной или азотной кислотами и предназначен для растительных и сливочных масел, маргарина, пищевых жиров и сыров.
Растворение проб.
Для раствор-я тв тел их обрабатывают мин. кислотами при нагревании или без него. Нередко исп-т смесь кислот (серной, азотной, соляной и др). Часто в к-ты добавляют окислители (пероксид водорода, бром и др). Подбор растворителя упрощается, если основные компоненты пробы известны из предварительных данных. Например, при определении сурьмы, мышьяка, германия избегают обработки исходной пробы соляной кислотой и стремятся не нагревать солянокислые растворы. Если же нельзя обойтись без нагревания, то анализ проводят с помощью обратного холодильника, чтобы не допустить потерь в-ва за счет его улетучивания в хлоридах.
Растворение производят в хим стаканах и конических колбах. Если при этом образуется пена или возможно разбрызгивание, то примен-ся установки, обеспечивающие улавливание пены или капель р-ра. Нередко используют колбы или тигли с затворами, кот позволяют выпускать образовавшиеся газы и защищают реакционную смесь от контакта с воздухом.
Если растворение протекает при t выше t кипения растворителя, то применяются запаянные ампулы или автоклавы.
Сплавление пробы.
При разложении некот материалов, например силикатных изделий, разл горных пород и др для полного растворения пробы одной обработки растворителя бывает недостаточно. В этих случаях для вскрытия пробы применяют ее сплавление с различными в-вами, котор. нызыв. плавнями. Например, щелочные плавни, т.к.карбонаты, пероксиды, а так же применяют кислотные плавни, т.к. гидросульфаты. При сплавлении происходит разложение анализируемого в-ва, нередко сопровождающееся окислением компонентов пробы кислородом воздуха. Чтобы усилить окислительные действия плава в него вводят нитраты, хлораты и др. окисл-ли. В этом случае протекает процесс спекания. Для усиления вводят нитраты, хлориды и др окислители. Получаемый после спекания/сплавления продукт растворяется в воде или в разбавл-х мин кислотах.
Пиролиз пробы.
Метод пиролитической газовой хроматографии широко применяется при исследовании полимерных материалов, пластмасс, каучуков, резин, лакокрасочных материалов и других высокомолекулярных соединений природного и искусственного происхождения. Метод основан на термическом разложении образцов пробы в инертной среде (без доступа кислорода) с последующим хроматографическим разделением и анализом образовавшихся летучих соединений. Таким образом, полученная пирограмма позволяет судить о составе исходного образца. Пиролиз применяется также при геохимических исследованиях.
При анализе орган-х материалов, а также для устранения орган-ских примесей из неорган-х проб применяется процесс пиролиза, в кот под действием высокой t происходит расщепление большинства молекул с образованием продуктов малой молек массы. По конечным или промежуточным процессам пиролиза устанавливают хар-ки исходных соединений. Пиролиз осущ-ся вспец печах с отводом продуктов расщепления при помощи потока инертного газа.
Сущность титриметрии
Титриметрический анализ основан на точном измерении объёма стандартного раствора реагента, израсходованного на реакцию с определяемым веществом. Расчёт результата титриметрии основан на принципе эквивалентности, в соответствии с которым вещества реагируют между собой в эквивалентных количествах. Его основными достоинствами являются точность, быстрота исполнения и возможность применения для определения самых разнообразных веществ. Определение содержания вещества в титриметрическом анализе осуществляется в результате проведения реакции точно известного количества одного вещества с неизвестным количеством другого, с последующим расчётом количества определяемого вещества по уравнению реакции. Основной операцией титриметрического анализа является титрование – постепенное смешивание веществ до полного окончания реакции. Обычно в титриметрическом анализе используются растворы веществ. В ходе титрования раствор одного вещества постепенно приливается к раствору другого вещества до тех пор, пока вещества полностью не прореагируют. Раствор, который приливают, называется титрантом, раствор, к которому приливается титрант, называется титруемым раствором. Объём титруемого раствора, который подвергается титрованию, называется аликвотной частью или аликвотным объёмом. Требования к реакциям в титровании:
1Реакция должна протекать количественно, т.е. константа равновесия реакции должна быть достаточно велика. Это означает, что реакция между определяемым веществом и реагентом должна протекать практически до конца в прямом направлении.
2Реакция должна протекать с достаточно большой скоростью.
3Реакция не должна осложняться протекание побочных реакций.
4Должен существовать достаточно точный способ определения окончания реакции.
Точка эквивалентности
ЭКВИВАЛЕНТ – некая реальная или условная частица числа Х, которая в данной кослотно-основной реакции эквивалентна 1 иону Н+ (реже иону ОН-) или в данной реакции окисления-восстановления эквивалентна 1 электрону.
Под условной частицей (структурным элементом) понимают как реально существующие частицы (электроны, ионы, атомы, молекулы), так и доли этих частиц, иногда их группы.
Фактор эквивалентности и эквивалент данного вещества являются непостоянными величинами, а зависят от стехиометрии реакции, определяющей число ионов водорода в реакции либо число полученных или отданных электронов в реакции.
Точка эквивалентности (в титриметрическом анализе) — момент титрования, когда число эквивалентов добавляемого титранта эквивалентно или равно числу эквивалентов определяемого вещества в образце. Точкой эквивалентности называется момент, наступающий в ходе титрования, когда реагирующие вещества полностью прореагировали. В этот момент они находятся в эквивалентных количествах, т.е. достаточных для полного, без остатка, протекания реакции.
Точка конца титрования
В точке эквивалентности наблюдается резкое изменение электропроводности.
Титрование проводят до точки конца титрования, когда с помощью приборн.методов устанавливают соответствие эквивалентности титранта и титруемого вещества. Точка конца титрования соответствует точному объему добавленного титранта, кот. не меньше эквивалент-го объема титранта, т.е. равно или чуть больше, эта точка устанавливается индикатор.методами, физико-хим.инструм-ми методами.
В индикаторных методах чаще всего исп.цветной индикатор, кот.изменяет свой цвет.
Кривые титрования
Кривая титрования – это график зависимости pH, оптической плотности или каких-либо других характеристик титруемого раствора (ось ординат) от объема добавленного титранта (ось абсцисс). Масштаб оси абсцисс всегда линейный, а оси ординат может быть линейным или логарифмическим. Линейный масштаб удобен для тех методов контроля за титрованием (спектрофотометрия, амперометрия), в которых контролируемый параметр меняется с концентрацией линейно, а логарифмический – в случае логарифмического изменения (например, при потенциометрии с ионоселективным электродом). Логарифмический масштаб часто используют при визуальном определении конечной точки титрования, поскольку именно в этом масштабе наиболее наглядно проявляется резкое изменение свойств раствора вблизи точки эквивалентности.
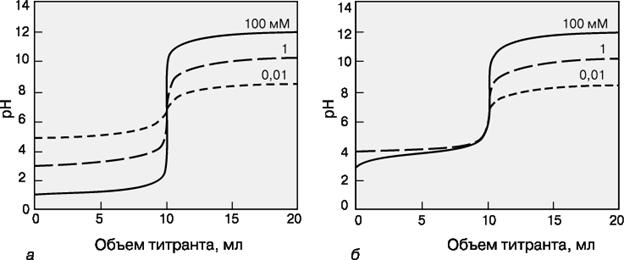 КРИВЫЕ ТИТРОВАНИЯ сильной кислоты сильным основанием (а) и слабой кислоты (pKa = 5) сильным основанием (б) для разных концентраций определяемого вещества.
КРИВЫЕ ТИТРОВАНИЯ сильной кислоты сильным основанием (а) и слабой кислоты (pKa = 5) сильным основанием (б) для разных концентраций определяемого вещества.
Способы титрования
В титриметрии испол. 3 способа титрования: прямое, обратное и по заместителю.
Прямое титрование – титров. р-ра опред.вещества А непосредственно р-м титранта В. Его применяют в том случае, если р-я между А и В протекает быстро. Содержание комп-та А при прямом титр-нии титрантом В рас-ся на основе р-ва п(А)=п(В). В кач-ве примера определ.в-ва сп-м прямого тит-я след.р-я титр-я:
 CH3COOH + NaOH CH3COONa + H2O
CH3COOH + NaOH CH3COONa + H2O
n (CH3COOH)=n (NaOH)
Обратное титр-е зак-ся в добавлении к опред.в-ву А избытка точно известного к-ва станд.р-ра В и после завершения р-ии между ними осущ.титров-е ост-гося кол-ва в-вар-м титранта В’. Этот способ применяют в тех случаях, когда р-я между А и В протек.нед.быстро, либо нет подх-гоиндикатора для фиксир-я точки экв-ти этой реакции. Пример:
 CaCO3 + 2HCl (из-к) CaCl2 + CO2 + H2O
CaCO3 + 2HCl (из-к) CaCl2 + CO2 + H2O
 HCl (ост) + NaOH NaCl + H2O
HCl (ост) + NaOH NaCl + H2O
n(1/2 CaCO3)=n(HCl)- n(NaOH)
Титрование по заместителю з-ся в титр-ии титрантом В не опред.в-ва А, а экв-го ему кол-ва з-ля А’, получив-гося в рез-те предвар.провед-й р-ии между опред.в-м А и каким-либо реагентом. Титров-е по з-лю примен.обычно в тех случаях, когда невозможно провести прямое титрование.
К-во молей эк-та опред-го в-ва титр-ии з-ля всегда равно к-ву молей эк-та титранта п(А)=п(В)=п(А’). пример:
 K2Cr2O7 + 6KI + 7H2SO4 Cr2(SO4)3 + 3I2 (з-ль) + 4K2SO4 + 7H2O
K2Cr2O7 + 6KI + 7H2SO4 Cr2(SO4)3 + 3I2 (з-ль) + 4K2SO4 + 7H2O
 I2 + 2Na2S2O3 2NaI + Na2S4O6
I2 + 2Na2S2O3 2NaI + Na2S4O6
n(K2Cr2O7)= n(I2)= n(Na2S2O3)
Гальванический элемент
Гальвани́ческий элеме́нт — химический источник электрического тока. Принцип действия гальванического элемента основан на взаимодействии двух металлов через электролит, приводящем к возникновению в замкнутой цепи электрического тока. Главные составные части гальванических элементов: два электрода разл. природы и электролит. Обычно электроды - это металлич. пластинки или сетки, на к-рые нанесены реагенты ("активные в-ва"); на отрицат. электрод - восстановитель (Zn, Li и др.), на положительный -окислитель (оксиды Mn, Hg и др., а также соли). На каждом из электродов, погруженных в электролит, устанавливается определенный потенциал (окислит.-восстановит. потенциал данной электродной р-ции); разность этих потенциалов в отсутствие тока наз. напряжением разомкнутой цепи (НРЦ). При соединении электродов между собой с помощью внеш. электрич. цепи электроны начинают перетекать от отрицат. электрода к положительному - возникает электрич. ток. Суммарная электрохим. р-ция на обоих электродах наз. токообразующей; по мере ее протекания восстановитель отдает, а окислитель присоединяет электроны. Ток прекращается при размыкании внеш. цепи, а также после израсходования запаса хотя бы одного из реагентов. ЭДС гальванического элемента зависит от материала электродов и состава электролита.
Метод прямой кондуктометрии
КОНДУКТОМЕТРИЯ - совокупность электро-хим. методов анализа, основанных на измерении электропроводности х жидких электролитов, к-рая пропорциональна их концентрации. Достоинства кондуктометрии: высокая чувствительность, достаточно высокая точность, простота методик, доступность аппаратуры, возможность исследования окрашенных и мутных р-ров, а также автоматизации анализа. Методы кондуктометрии бывают постояннотоковые и переменнотоковые последние могут быть низкочастотными или высокочастотными. В прямой кондуктометрии непосредственно определяют концентрацию эл-в в растворе. Метод применяется гл. обр. для анализа разб. р-ров. В случае концентрир. р-ров необходимо строить градуировочные графики. Опред-ся в присутств.других электролитов возможно, если концентрации последних постоянны. На методе прямой кондуктометрии основаны конструкции солемеров и др. кондуктометрич. устройств, позволяющих определять олеум, а также разл. соли в минеральной, речной и морскойводах, физиол. жидкостях и др. Прямую кондуктометрию применяют при контроле регенерации ионитов, очистки воды, промывки осадков, при оценке качества вин, соков и др. напитков, чистоты орг. р-рителей, газов, твердых солей, текстильных материалов, бумаги, зерна, почвы и т.д. Часто анализируемые образцы предварительно сжигают, а выделяющиеся газы поглощают подходящими р-рами. По электропроводности поглотителей определяют кол-ва газов (в частности, СО2, NO2, SO2), следовательно-содержание соответствующих элементов, напр. С, N, S, в металлах, сплавах и орг. соединениях.
Ионоселективные электроды
Ионоселективные электроды представляет собой устройство, основным элементом которого является мембрана, проницаемая только для определенного иона. Ионселективные электроды имеют следующие достоинства: они не оказывают воздействия на исследуемый раствор, портативны, пригодны как для прямых определений, так и в качестве индикаторов в титриметрии.Ион-е, электрохим. электроды, равновесный потенциал к-рых в р-ре электролита, содержащем определенные ионы, обратимо и избирательно зависит от концентрации этих ионов. На этом основании ион-ые электроды используют для определения концентрации (активности) разл. ионов в р-ре, а также для анализа и контроля процессов, протекание к-рых сопровождается изменением ионного состава р-ров. Разработка и применение ион-ых электродов для определения разл. ионов - осн. задача потенциометрии. В большинстве случаев ион-ый электрод представляет собой устройство, осн. элементом к-рого является мембрана, проницаемая только для определенного иона. Между р-рами электролитов, разделенных мембраной, устанавливается стабильная разность потенциалов, к-рая алгебраически складывается из двух межфазных скачков потенциала и диффузионного потенциала, возникающего внутри мембраны.
Типы:
1. первичные:
- электроды со стеклянной мембраной
- кристаллические мембранные электроды – мембраны состоят из ионных кристаллов или их смесей. Иногда представляют собой монокристаллы.
- электроды с жидкой мембраной – основу мембраны составляют несмешивающиеся с водой жидкости. Внутренний р-р сравнения отделён от анализируемого р-ра тонким слоем жидкости содержащий жидкий ион, не смешивающийся с водой, но селективно реагирующий с определённым ионом: жидкие иономембранные электроды и электроды с жидкой мембраной с нейтральным носителем
2.сложные и многомембранные
- молекулярно-чувствительные устройства, такие как газочувствительные или ферментные электроды, в которых потенциометрический детектирующий блок основан на стандартных потенциометрический электродах ранее перечисленных типов.
Прямая кулонометрия
При прямой кулонометрии, определяют в-во окислившееся либо восстановившееся на одном из электродов, при этом побочные хим р-ции не протекают. Этот метод прямой кулонометрии примен для опред многих орг в-в, при этом различают на практике кулонометрию при постоян потенциале и при постоян силе тока.
Кулонометрия при пост потенц (прям кулонометрия): В этом случае при пост потенциале эл-за сила тока пост изменяется, т.к. конц определяемого в-ва непрерывно падает. Сила тока измен-ся по экспоненте в зависимости от времени эл-за, если построить графич зависимость в корд (ln I от t), то зависимость – прямая линия. Ln I=kt: k=tg α.
Схема прямой кулонометрии при пост потенц. 1 – источ питания, 2 – реастат, 3 – амперметр, 4- раб эл-д, 5 – эл-д, 6 – стандарт эл-д, 7 – потенциом электролизер.
Кулонометрия при пост силе тока (хронометр. М-д): При этом м-де сила тока постоянная величина, а продолжительность электролиза t опред секундомером, затем рассчит Q: Q=I*t. Постоянную силу тока поддерживают включая последовательно с электролитической ячейкой высокополное сопротивление порядка 1*104 – 2.5*10 4. Наряжение поддерживается 100-200 В. Напряжение на ячейках обычно повышается при электролизе вследствие изменения концентрации ионов водорода в растворе. Схема: 1 – источ пит, 2 – ключ, 3 – реастат, 4 – амперметр, 5 – яч для эл-за.
Методы полярографии
Методы полярографии. 1.Метод градуировочного графика. При этом методе снимают несколько полярограмм стандартных образцов, по высоте Н и концентрации С строят градуировочный график, чаще всего это прямая проходящая от начала координат: Н = f(C). По высоте волны Н неизвестного образца определяют его концентрацию про градуировочному графику.
2. Метод стандарта. В это методе присутствуют строго определенные условия, в которых снимают полярограммы стандартного и анализируемого раствора. Концентрацию неизвестного раствора находят по следующей формуле: Сх=Сст* (Нх/Нст), где Нх,ст – высота волны, искомого раствора и стандартного. Сст – концентрация стандартного растворо. 3. Метод добавок. В этом методе учитывают линейный характер зависимости Iх = к*Сх. Если в раствор добавить известное количество стандартного раствора то сила тока изменяется и станет равной в соответствии с этой зависимостью Iх+ст = к(Сх+Сст). Сх определяют, поскольку Сст известно, а Iх.
Дозатор
Колонка
Дектор
Усилитель
Регистрирующее устройство
Область термостатирования

50. Хромотографич колонки: Колонки – трубки, наполнен опред сорбентом. Трубка из нержав стали, меди, стекла. Сорбенты: Активир уголь, селикогели, оксид алюминия, спец жид нанесен на поверх малоактив адсорбента.Такие колонки – насадочные. Исп также капиллярные колонки, предст собой группу с внутр диаметром = 0,2 мм, а жидкость нанос непосредств на внутр поверх капилляр трубки. Применение того или иного типа колонок и выбор наполнителя зависит от хим состава анализир смеси.
В хроматографической колонке происходит разделение исходной многокомпонентной смеси на ряд бинарных смесей, состоящих из жидкости или газа-носителя (для газовой хроматографии) и одного из разделяемых компонентов. Разделение может происходить как за счет образования временных связей между разделяемыми веществами и неподвижной фазой колонки (адсорбционная хроматография), так и по иным принципам (например, эксклюзионная хроматография). Точнее значения объема или времени выхода каждого компонента из колонки устанавливают при калибровке.
51. Детекторы хроматографические. Устройства для количеств.и качеств. определения веществ разделяемой смеси в потоке подвижной фазы на выходе из хроматографич. колонки. Детекторы можно рассматривать также как преобразовательный элемент, в котором изменение состава проходящей через него смеси преобразуется в изменение выходного сигнала. Различают детекторы дифференциального и интегрального типа. Первые регистрируют мгновенное значение одной из характеристик (концентрации или потока), вторые суммируют кол-во вещества за определенный промежуток времени. Детектор по теплопроводности (ДТП) К универсальным относится детектор по теплопроводности (ДТП, устаревшее и нерекомендованное название — катарометр). Принцип его действия заключается в изменении температуры нагретой нити при обдувании её газом (пробой) с разной теплопроводностью. Пламенно-ионизационный детектор (ПИД) Этот детектор селективно определяет углеводороды. Принцип его действия заключается в изменении силы тока в плазме водородно-кислородного пламени при попадании в неё горючих соединений углерода. Электронозахватный детектор (ЭЗД) В данном виде детектора используется источник бета-частиц (электронов), как правило, 63Ni, или альфа-частиц (269Pu). Если в газе, проходящем мимо такого радиоактивного источника, оказываются молекулы, склонные к ионизации, возникает пропорциональный их концентрации ток, который можно измерить. Своеобразной разновидностью электронозахватного детектора является детектор дифференциальной ионной подвижности (ДДИП), весьма компактный и поэтому доступный для использования в портативных хроматографах.
54. ИЗОТOПНОГО РАЗБАВЛЕНИЯ МЕТОД, метод количеств. хим. анализа с использованием радиоактивных или обогащенных стабильных нуклидов в качестве индикаторов. Основан на изменении изотопного состава определяемого элемента в результате разбавления при смешении с анализируемым образцом. Особенность метода - возможность проводить количеств. определения при неполном выделении в-ва. В классич. варианте изотопного разбавления метода определение компонента основано на изменении удельной радиоактивности (S) при разбавлении в ходе анализа.
Абсолютная и относительная погрешности.
Абсолютная погрешность – отклонение результата измерения от истинного значения измеряемой величины: ΔXͥi=Xi-µ, где Xi – измеряемая величина, µ - истинное значение измеряемой или анализируемой величины.
В большинстве случаев µ неисвестно, и абсолютная погрешность не может быть точно определена, поэтому часто определяют верхнюю границу ΔXͥi.
Предельная абсолютная погрешность ΔX приближенно ΔXͥi является наименьшим числом: ΔX≥l ΔXͥil, тогда интервал, в котором заключено точное значение числа µ будет находится в этих пределах:
Х- ΔX≤µ≤ Х+ΔX, где Х – измеряемая величина.
Абсолютные погрешности не полностью характеризуют отклонение величины от истинного значения.
ΔX=1, но в первом случае µ1=10, а во втором µ2=1000.
Поэтому используют понятие относительной погрешности (ошибки), кот представ собой отношение абсолют погрешности к истинному значению измеряемой величины: Ɛ= ΔX/µ.
Обычно относительная погрешность измер в %: Ɛ= ΔX/µ*100%.
2 Классификация погрешностей по форме представления:
- абсолютна я - отклонение результата измерения от истинного назначения измеряемой величины. ∆Xi=Xi-µ
- относительная - отношение абсолютной погрешности к истинному значению. E=∆X/µ
- приведённая - погрешность, выраженная отношением абсолютной погрешности средства измерений к условно принятому значению величины, постоянному во всем диапазоне измерений или в части диапазона. E =∆X/ µn, где µn - нормирующее значение, которое зависит от типа шкалы измерительного прибора и определяется по его градуировке.
Погрешности измерений и анализа по характеру проявлений условно подразделяют на 3 группы: Систематические Случайные Грубые
Систематические – погрешности, которая при повторных измерениях остается постоя
|
|
|

Особенности сооружения опор в сложных условиях: Сооружение ВЛ в районах с суровыми климатическими и тяжелыми геологическими условиями...

Механическое удерживание земляных масс: Механическое удерживание земляных масс на склоне обеспечивают контрфорсными сооружениями различных конструкций...
Индивидуальные и групповые автопоилки: для животных. Схемы и конструкции...
Наброски и зарисовки растений, плодов, цветов: Освоить конструктивное построение структуры дерева через зарисовки отдельных деревьев, группы деревьев...
© cyberpedia.su 2017-2024 - Не является автором материалов. Исключительное право сохранено за автором текста.
Если вы не хотите, чтобы данный материал был у нас на сайте, перейдите по ссылке: Нарушение авторских прав. Мы поможем в написании вашей работы!