Раздел 2. ЛАБОРАТОРНЫЕ РАБОТЫ
Прямая потенциометрия (ионометрия).
Определение нитратов в овощах
Цель работы: освоение техники потенциометрических измерений, способов определения параметров ионоселективных электродов, способов нахождения концентрации методом прямой потенциометрии, методики определения нитратов в пищевых продуктах.
Введение
В данной работе в качестве индикаторного электрода используется мембранный нитрат-селективный электрод, в качестве электрода сравнения – хлорсеребряный. Для такой системы электродный потенциал (Е) определяется активностью ионов NO3–:
E = const – ϑ lg a(NO3–),
где ϑ = 2,303∙R∙T/F. Теоретически ϑ = 59 мВ при 25 оС. По паспорту используемого ионоселективного электрода наклон градуировочной функции составляет (55 ± 4) мВ.
В присутствии инертного (фонового) электролита и при постоянной температуре ионная сила растворов постоянна, следовательно, коэффициент активности также постоянен, потенциал электрода связан с концентрацией нитрат-иона:
E = const/ – ϑ lg[NO3–] = const/ + ϑ pNO3
В качестве фонового электролита при определении нитрат-иона в реальных объектах анализа обычно используется 1%-ный раствор алюмокалиевых квасцов.
Экспериментальная часть
Оборудование: анализатор жидкости ЭКСПЕРТ–001; нитрат-селективный и хлорсеребряный электроды; магнитные мешалки.
Реактивы: раствор KNO3 1 М; раствор KCl 0,1 М; раствор K2SO4 0,1 М; раствор СН3СООNa 0,1 М; раствор KAl(SO4)2·12H2O 1%-ный; анализируемые овощи (картофель или морковь).
Подготовка оборудования к работе и техника проведения измерений: Анализатор жидкости ЭКСПЕРТ – 001 предназначен для измерения активности (рН, рХ) и массовой концентрации ионов (С), окислительно-восстановительного потенциала (Eh) в питьевых, природных, сточных водах и водных растворах проб растительной, пищевой продукции, почв и т.д. Кроме того, анализатор может использоваться в качестве высокоомного милливольтметра при потенциометрическом титровании, проведении измерений методом стандартных добавок.
Электроды, необходимые для проведения измерений, подготавливают в соответствии с их паспортами. Несоблюдение правил эксплуатации и хранения, неаккуратное и небрежное обращение с электродами ведет к их порчи. Помните, что электроды – невосстанавливаемое и дорогое изделие!
Подключите индикаторный электрод (нитрат-селективный) к разъему «ИЗМ», электрод сравнения (хлорсеребряный) – к разъему «ВСП», установите электроды в штатив и поместите их в лабораторный стакан с дистиллированной водой. Включите прибор в сеть (кнопка «ВКЛ»). Прибор должен прогреться в течение 15 минут.
Электроды перед погружением в каждый раствор необходимо тщательно промывать дистиллированной водой. Для этого, осторожно поднимите штатив с электродами вверх, следя за тем, чтобы электроды не стукались о стенку стаканчика, а сам стаканчик не опрокинулся. Для промывки электродов необходимо ополоснуть их в большом количестве дистиллированной воды (удобно пользоваться промывалкой). Осушите (осторожно) электроды фильтровальной бумагой, ополосните электроды и измерительную ячейку небольшой порцией раствора, в котором будут проводиться измерения, залейте в стакан необходимое количество исследуемого раствора и аккуратно опустите в него электроды. Электроды должны быть погружены в перемешиваемый раствор как минимум на три сантиметра. Следите за тем, чтобы на измерительной части электрода не было пузырьков воздуха.
В большинстве экспериментов данной лабораторной работы проводят измерение ЭДС электродной системы, для этого прибор переводят в режим « Вольтметр (Eh)» кнопками «◄» «►». На дисплее должна появиться надпись:
Выбор режима
Вольтметр (Eh)
Нажмите кнопку «ИЗМ». На дисплее появится надпись:
Вольтметр 00:02
ххх,х мВ
Начнется измерение ЭДС и отсчет времени измерения. В течение 1 – 2 минут отметьте установившееся значение ЭДС. Для выхода из режима измерений нажмите кнопку«ОТМ» и только после этого поменяйте анализируемый раствор.
Ход работы
Обработка результатов
1. Рассчитайте активность нитрат-ионов в приготовленных растворах с учетом ионной силы. По полученным значениям постройте зависимость E– lg а(NO3-), рассчитайте по МНК коэффициенты а и b, отвечающие нернстовскому участку прямой, определите угловой коэффициент этого участка.
2. Оцените нижнюю границу линейности градуировочной функции:
- по значению lg а(NO3-), которому отвечает точка пересечения экстраполированных линейных участков (наклонного и горизонтального);
- по значению а(NO3-), которому соответствует равное (ϑ/z)∙lg2 отклонение экспериментальной кривой от экстраполированного нернстовского линейного участка.
Опыт 2. Определение коэффициента селективности ионоселективного электрода
Из 1 М раствора KNO3 приготовьте последовательным разбавлением растворы KNO3 с концентрациями 1∙10-1; 1∙10-2; 1∙10-3; 1∙10-4; 1∙10-5; 1∙10-6; 1∙10-7 моль/л. Растворы доводите до метки в мерных колбах 0,1 М раствором соли, содержащей мешающий анион (КСl, K2SO4 или CH3COONa по указанию преподавателя). Проведите измерения потенциала в приготовленных растворах, начиная с наименьшей концентрации KNO3, как в опыте 1.
Обработка результатов
1. По полученным данным постройте график зависимости E – lg C(NO3-).
2. Экстраполируя линейный участок кривой (при высоких концентрациях NO3--иона) и участок, где потенциал электрода практически не зависит от C(NO3-), до пересечения, найдите значение lg C(NO3-), при котором потенциал нитрат-селективного электрода в равной мере определяется основным и мешающим ионами.
Рассчитайте коэффициент селективности:

где k х,yпот – потенциометрический коэффициент селективности мембраны к мешающему иону; a x – активность определяемого иона; z x – его заряд; a y – активность мешающего иона; z y – заряд мешающего иона.
3. Найдите значение a х, которому соответствует отклонение экспериментальной кривой от экстраполированного нернстовского участка, равное (ϑ/ z х)∙lg2. Рассчитайте коэффициент селективности.
Обработка результатов
1. Постройте градуировочную зависимость в координатах Е (мВ) – lgC(NO3-) и найдите С(NO3-) в анализируемом растворе.
2. Рассчитайте по МНК уравнение линейного участка графика,найдите С(NO3-) в анализируемом растворе.
3. Рассчитайте концентрацию нитрат-иона в анализируемом растворе по методу добавок.
4. Найдите концентрацию нитрат-иона в анализируемом растворе по результатам измерений рХ.
5. Рассчитайте массу нитрата в пробе (мг) и содержание нитрата в пробе (мг/кг) из концентраций, найденных каждым из использованных Вами способов; сравните с ПДК на нитраты, мг/кг: картофель – 250, морковь ранняя – 400, морковь поздняя – 250.
Дополнительная литература
1. ГОСТ 29270-95. Продукты переработки плодов и овощей. Методы определения нитратов.
2. Скурихин И.М., Нечаев А.П. Все о пище с точки зрения химика. – М.: Высшая школа, 1991.
Введение
Определение марганца основано на потенциометрическом титровании по методу окисления-восстановления ионов Mn2+, полученных при растворении навески руды или стали, стандартным раствором перманганата калия в нейтральной пирофосфатной среде. При титровании индикаторным электродом служит гладкий платиновый электрод, а электродом сравнения – насыщенный хлорсеребряный. В растворе происходит реакция:
3 Mn2+ + 2 MnO4- + 2H2O = 5 MnO2 + 4H+
Одним из промежуточных продуктов этой реакции является ион Mn3+, неустойчивый в отсутствие стабилизирующих его лигандов и диспропорционирующий по уравнению:
2 Mn3+ + 2H2O = 5 MnO2 + Mn2+ + 4H+
При введении фторида аммония или кислого пирофосфата натрия образуются прочные комплексы, и Mn3+ становится устойчивым. Так, в нейтральной среде Mn2+ окисляется перманганатом не до диоксида марганца, а до пирофосфатного комплекса Mn3+:
4 Mn2+ + MnO4- +15 H2P2O72- + 8H+= 5 [ Mn(H2P2O7)3]3- + 4H2O
Поскольку комплекс Mn3+ с пирофосфатом окрашен, то определение точки эквивалентности с индикатором затруднено, и применяют потенциометрическое титрование.
До точки эквивалентности электродный потенциал определяется равновесием:
Mn2+ + 3 H2P2O72- – ē = [ Mn(H2P2O7)3]3-
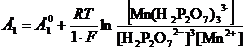
После того, как все ионы Mn2+ будут окислены перманганатом, возникает скачок потенциала из-за появления избытка перманганата, равновесие на электроде:
MnO4- + 3 H2P2O72- + 8H+ + 4ē = [ Mn(H2P2O7)3]3- + 4H2O

Комплекс Mn3+ с пирофосфатом неустойчив в кислой среде, поэтому титрование ведут в нейтральном растворе. Увеличению скачка способствует большое значение равновесной концентрации пирофосфат ионов, а повышение температуры ускоряет реакцию.
В лабораторной работе титрование проводится до постоянного значения потенциала с использованием блока автоматического титрования БАТ-15. Предварительно, при титровании в ручном режиме, определяют потенциал, соответствующий точке эквивалентности.
Экспериментальная часть
Оборудование: рН-метр-милливольтметр рН-121; блок автоматического титрования БАТ-15 с электромагнитным клапаном; платиновый электрод; хлорсеребряный электрод; микробюретка с запасным резервуаром на 2 мл; мешалка; плитка.
Реактивы: растворы HNO3 1:1, 1 н.; раствор HCl 1:1; раствор NaOH 1 н.; раствор KMnO4 ~0,002 М; Na4P2O7·10H2O (крист.); индикаторная бумага.
Подготовка оборудования к работе и техника проведения измерений: Потенциометр рН-121 (рН-метр-милливольтметр) служит для точного измерения малых ЭДС, например, ЭДС, создаваемых электродными системами. Его шкала проградуирована в единицах рН (соответствует сотням милливольт), а также в °С. Прибор рН-121 обычно используется в одном из двух режимов: измерение рН со стеклянным электродом, имеющим водородную функцию, и измерение ЭДС. Каждый из этих режимов может использоваться для контроля точки эквивалентности в соответствующих методиках потенциометрического титрования.
При работе прибора с электродными системами в любом из вышеуказанных режимов электроды должны быть заранее подготовлены. При неправильной эксплуатации электроды быстро выходят из строя. Помните, что электроды – невосстанавливаемое и дорогое изделие! Подключите индикаторный электрод (платиновый) к разъему «ИЗМ». Будьте внимательны, поскольку в приборе два разъема под индикаторные электроды, которые переключаются кнопкой «ИЗМ1/ИЗМ2» на передней панели прибора (при нажатии включается второй разъем). Электрод сравнения (хлорсеребряный) подключите к разъему «ВСП», установите электроды в штатив и поместите их в лабораторный стакан с анализируемым раствором.
Включите прибор в сеть (тумблер сбоку). При наличии напряжения на передней панели загорается контрольная лампочка. Прибор должен прогреться в течение 30 минут.
При замене растворов, промывке электродов, хранении прибора – электродная система всегда, кроме непосредственных измерений на приборе, должна быть отключена. Для этого нужно нажать черную кнопку «0, t». Эту кнопку отжимают (аналогия с арретиром весов) только во время измерений по шкале.
Электроды перед погружением в каждый раствор необходимо тщательно промывать дистиллированной водой. Платиновый электрод можно погрузить на 1 – 2 секунды в раствор HNO3 (1:1), затем тщательно промыть дистиллированной водой. Осушите (осторожно) электроды фильтровальной бумагой, залейте в стакан необходимое количество исследуемого раствора и аккуратно опустите в него электроды. Электроды при этом должны быть погружены в перемешиваемый раствор как минимум на три сантиметра.
Для измерения ЭДС с помощью рН-121 сначала выбирают широкий диапазон значений -1÷14 (нижняя шкала) и нажимают кнопку «mV», при этом кнопка «0, t» автоматически отжимается. В случае зашкаливания стрелки в отрицательную область нажмите «–mV» и измеренным значениям ЭДС припишите знак минус. Для более точного измерения ЭДС выбирают один из узких диапазонов: -1÷4, 4÷9 или 9÷14 (верхние шкалы, самая верхняя шкала для учета температуры раствора). Запишите уточненное значение ЭДС, умножая показания шкалы на 100 и не забывая о полярности. Не забывайте также при смене раствора и промывке электродов отключать электродную систему кнопкой «0, t».
Ход работы
Опыт 1. Приготовление анализируемого раствора
1. Навеску стали (~ 1 г) растворите при нагревании в конической колбе на 100 мл в 20 мл смеси разбавленных кислот соляной (1:1) и азотной (1:1).
2. После растворения навески к раствору осторожно прибавьте несколько капель концентрированной HNO3 для окисления железа (II) и затем кипятите до полного удаления оксидов азота и избытка кислоты, но не досуха. Если все же выпарили досуха, то прибавьте 5-7 мл НCl (1:1) и вновь прокипятите.
3. Полученную жидкость перенесите в мерную колбу на 250,0 мл и доведите водой до метки, не отфильтровывая выпавший осадок (возможно образование осадка кремниевой кислоты).
4. В стакан для титрования (на 100 мл) налейте мерным цилиндром 25 мл 10%-ного теплого свежеприготовленного раствора пирофосфата натрия и прибавьте к нему мерной пипеткой при перемешивании 25,0 мл анализируемого раствора. Образующийся вначале белый осадок растворяется. Полученный раствор нейтрализуйте по индикаторной бумаге, добавляя несколько капель 1 н. раствора NaОН или 1 н. раствора HNO3 в зависимости от реакции среды.
Опыт 2. Потенциометрическое титрование в ручном режиме
Оттитруйте анализируемый раствор вручную раствором перманганата калия, измеряя значения потенциала через минуту после прибавления титранта. Вначале проведите грубое титрование, прибавляя по 0,1 – 0,2 мл титранта из микробюретки. Затем титрование повторите с новой аликвотой анализируемого раствора, вводя титрант в области скачка порциями по 0,05 мл.
Обработка результатов
На основании полученных данных точного титрования постройте кривые титрования в координатах Е = f(V), ΔЕ/ΔV = f(V) и Δ2Е/ΔV2 = f(V); найдите объемKMnO4 в конечной точке титрования, и соответствующее значение Ектт – его далее задают в качестве конечной точки при автоматическом титровании.
Опыт 3. Потенциометрическое титрование в автоматическом режиме
Проведите автоматическое титрование аликвот анализируемого раствора с помощью блока автоматического титрования БАТ-15. Рекомендуемые параметры: время выдержки 45 с; ширина зоны 2 ед. рН (200 мВ). Получите не менее 3 результатов параллельных титрований.
Блок автоматического титрования БАТ-15 предназначен для проведения полуавтоматического потенциометрического титрования в комплекте с бюреткой и рН-метром-милливольтметром (рис. 1).

Рис. 1. Функциональная схема установки для титрования.
Напряжение Ux, пропорциональное ЭДС электродной системы, с выхода рН-метра-милливольтметра подается на вход БАТ-15, где сравнивается с напряжением UK, установленным на задатчике конечной точки титрования. Разность этих напряжений поступает на вход усилителя. На выходе усилителя включено бесконтактное электронное реле, управляющее работой электромагнитного клапана. Клапан открывает или закрывает подачу титрующего раствора из сосуда в ячейку с титруемым раствором. При Ux = UК реле отключает питание клапана, который, пережимая резиновую трубку, прекращает подачу титрующего раствора.
1. Рекомендуется рН-метр-милливольтметр устанавливать на блоке автоматического титрования, а штатив с магнитной мешалкой и микробюретку справа от приборов. Соедините гнезда «2V» рН-метра-милливольтметра с гнездами «ВХОД, 0…2V» блока автоматического титрования специальным кабелем. Подсоедините электромагнитный клапан к БАТ. Блок титрования прогрейте в течение 30 минут (кнопка «СЕТЬ»).
2. Закрепите клапан на штативе таким образом, чтобы он находился ниже уровня крана бюретки. Выдвиньте задвижку на клапане и установите в паз резиновую трубку, затем задвижку установите на место. Заполните микробюретку раствором и верхний конец резиновой трубки соедините с микробюреткой. Откройте кран микробюретки. Заполните капилляр раствором титранта, для этого удерживайте кнопку «РУЧН».
3. Установите диапазон измерений рН-метра-милливольтметра, на котором будет вестись титрование (широкий от минус 1 до 14 или узкий с размахом 5 ед. рН). Установите на задатчике «ЗАДАННАЯ ТОЧКА» значение конечной точки титрования в единицах рН (значение Е ктт, мВ, деленное на 100). При работе рН-метра на широком диапазоне значение ктт определяется суммой значений, соответствующих нажатой кнопке БАТ «–1, 4 или 9», и показаний реохорда задатчика «ЗАДАННАЯ ТОЧКА». При работе прибора на одном из узких диапазонов с размахом 5 рН ктт определяется как сумма значения начала диапазона рН-метра-милливольтметра и установленного значения по реохорду задатчика. При этом должна быть нажата кнопка «–1, УЗКИЙ».
4. Установите кнопку «ВВЕРХ-ВНИЗ» в одно из положений в зависимости от характера проводимого титрования. Кнопка отжата («ВВЕРХ») при титровании раствора до более высоких значений рН или ЭДС. Кнопка нажата («ВНИЗ») при титровании до более низких значений.
5. Ручкой «ВЫДЕРЖКА» установите необходимую величину выдержки. Вследствие малой скорости протекания реакции, протекания вторичных процессов, перемешивания растворов и пр. может происходить изменение потенциала (рН) после прекращения подачи раствора. В этом случае прибор позволяет проводить дотитрование раствора в пределах установленного значения времени выдержки. Поэтому при наличии выше указанных факторов выдержку установите в пределах 25 – 45 с.
6. Установите ручкой «ЗОНА» выбранную ширину зоны импульсной подачи раствора титранта. Ширина зоны не должна быть равна нулю. Оптимальная установка ширины зоны импульсной подачи раствора зависит от формы кривой титрования. Когда кривая титрования имеет крутой наклон вблизи эквивалентной точки (например, при титровании сильной кислоты сильной щелочью), установите ширину зоны по возможности большей. В случае пологой кривой в целях экономии времени используйте узкую зону импульсной подачи раствора (т.е. установите ручку «ЗОНА» на значении не больше 2).
7. Для автоматической блокировки клапана во время титрования и необходимой выдержки нажмите кнопку «ВКЛ».
8. Положение капилляра относительно измерительного электрода значительно влияет на скорость и точность титрования. Если кривая имеет крутой фронт, капилляр располагают на некотором расстоянии от измерительного электрода, чтобы титрование вблизи точки эквивалентности проходило плавно. В данном случае очень важно перемешивание. При пологом фронте кривой титрования капилляр расположите близко к измерительному электроду, чтобы избежать перетитровывания раствора.
9. Начните процесс титрования нажатием кнопки «ПУСК». При этом загорается лампочка «ПРОЦЕСС», и начинается подача раствора. Отсчет количества титрующего раствора следует производить по истечении установленной выдержки, т.е. после загорания лампочки «КОНЕЦ». После окончания титрования отожмите кнопку «ПУСК», чтобы избежать выхода из строя регулирующего клапана.
10. Повторите титрование. В случае перетитровки или недотитровки раствора переместите ручку задатчика «ЗАДАННАЯ ТОЧКА» в меньшую или большую сторону.
11. После окончания работы с титратором кран бюретки перекройте, а капилляр промойте.
Обработка результатов
Необходимую для расчетов молярную концентрацию раствора перманганата калия уточните у лаборанта. Нормальную концентрацию этого раствора найдите с учетом фактора эквивалентности (см. уравнение реакции титрования). По закону эквивалентов рассчитайте результат анализа – массовую долю марганца в образце стали.
Экспериментальная часть
Оборудование и посуда: анализатор жидкости ЭКСПЕРТ–001; стеклянный и хлорсеребряный электроды; магнитная мешалка; бюретка на 25 мл.
Реактивы: стандартный раствор HCl 0,1000 M (по фиксаналу); раствор KOH ~ 0,2 М; контрольный раствор – смесь соляной и уксусной кислот; анализируемый фруктовый сок.
Подготовка оборудования к работе и техника проведения измерений: Подготовка анализатора жидкости ЭКСПЕРТ-001 и техника работы с электродами описаны в работе 2.1. В качестве индикаторного электрода используется стеклянный электрод. Для измерения рН сначала необходимо провести калибровку прибора по стандартным буферным растворам, этим предварительно занимается лаборант или инженер. Данные вносятся в память анализатора.
Для измерения рН анализируемого раствора кнопками «◄» «►» установите режим «рН-метр-иономер».На дисплее появится надпись:
Выбор режима
рН-метр-иономер
Проверьте правильность выбора иона. Для этого, нажмите кнопку «ИОН», на дисплее должна появиться надпись:
рН
В случае отображения другой информации кнопками «v» «w» выберите необходимый пункт – рН.
Нажав кнопку «ВВОД», вы подтвердите правильность выбора иона и снова выйдете в режим «рН-метр-иономер».
Для проведения измерений нажмите кнопку "ИЗМ". На дисплее появится надпись:
рН 00:02
х,хx pX
Начнется измерение рН и отсчет времени. Запись значений проводят в течение одного и того же интервала времени (30 – 60 с).
Для выхода из режима измерения нажмите кнопку "ОТМ". Вы снова выйдете в режим "рН-метр-иономер". Промойте электроды дистиллированной водой и подготовьте следующий раствор.
Ход работы
Опыт 1. Стандартизация раствора гидроксида калия по соляной кислоте
В ячейку для титрования отберите аликвоту 10,0 мл стандартного раствора HCl. Бюретку заполните раствором КОН. Включите магнитную мешалку и проведите титрование, записывая объемы добавленного титранта и соответствующие значения рН. В области скачка титрования (VТЭ ± 1,5 мл) добавляйте титрант порциями по 0,2 мл; вне области скачка титрования – по 1 мл. Титрование ведите до прибавления значительного избытка титранта (до рН 12 – 13). Повторите титрование до получения 3 параллельных результатов.
Обработка результатов
1. Постройте кривые титрования (отдельно для каждого из параллельных титрований):
- интегральную (рН – V)
- дифференциальную по первой производной ( – V)
– V)
- дифференциальную по второй производной (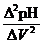 – V)
– V)
- по первому методу Грана ( – V)
– V)
- по второму методу Грана (G – V, гдеG = (Vo + V)10-pH на участке до точки эквивалентности (ТЭ) и G = (Vo + V)10pH на участке после ТЭ)
2. Определите в каждом случае графически конечную точку титрования (КТТ).
3. Рассчитайте по результатам титрований точную концентрацию раствора КОН, сопоставьте результаты, полученные разными способами.
Опыт 2. Определение соляной и уксусной кислот в их смеси
1. Получите у лаборанта стакан с порцией контрольного раствора – смеси соляной и уксусной кислот. Включите магнитную мешалку и проведите грубое титрование, добавляя по 1 мл титранта и записывая объемы добавленного титранта и соответствующие значения рН. Титрование ведите до достижения рН ~12. Найдите области двух скачков титрования – те значения V т, при которых добавление 1 мл титранта вызывает наиболее сильное изменение рН.
2. Проведите точное титрование другой порции контрольного раствора. Добавляйте титрант по 1 мл, а в области скачков титрования – по 0,2 мл. Внесите данные в таблицу. Повторите титрование до получения 3 параллельных результатов.
Обработка результатов
1. Постройте интегральные кривые по данным точного титрования (отдельно для каждого из параллельных титрований).
2. Для определения КТТ используйте три различных способа, обоснуйте свой выбор.
3. Рассчитайте массу каждой из кислот в контрольном растворе. Получите у лаборанта действительные значения масс кислот и рассчитайте относительную ошибку своего результата.
Опыт 3. Определение кислотности фруктового сока
Общая (титруемая) кислотность – один из нормируемых показателей качества многих видов продуктов. Так, согласно федеральному закону № 178-ФЗ от 27.10.2008 «Технический регламент на соковую продукцию из фруктов и овощей»:
· массовая доля титруемых кислот в соковой продукции из фруктов и (или) овощей для детей раннего возраста должна составлять не более 1,2 процента для соковой продукции из цитрусовых фруктов (в пересчете на безводную лимонную кислоту) и не более 0,8 процентов для соковой продукции из других видов фруктов, овощей (в пересчете на яблочную кислоту);
· массовая доля титруемых кислот в соковой продукции из фруктов и (или) овощей для детей дошкольного и школьного возрастов должна составлять не более 1,3 процента (для соковой продукции из цитрусовых фруктов в пересчете на безводную лимонную кислоту, для соковой продукции из других видов фруктов, овощей в пересчете на яблочную кислоту).
1. В ячейку для титрования отберите мерной пипеткой аликвоту 10 – 25 мл анализируемого сока. Включите магнитную мешалку и проведите грубое титрование. Найдите область скачка титрования.
2. Проведите точное титрование 3 аликвот сока такого же объема.
Обработка результатов
1. Постройте интегральные кривые титрования.
2. Определите КТТ тремя различными способами.
3. Рассчитайте массовую долю (%) преобладающей в данном продукте кислоты (табл. 1), приняв плотность жидкости равной 1,0 г/мл.
Таблица 1
Органические кислоты, преобладающие в плодах и ягодах
| Органическая кислота
| Продукты
|
| Название
| Формула
| f экв
|  , г/моль , г/моль
|
| яблочная
| HOOCCH(OH)CH2COOH
| 
| 67,04
| семечковые и косточковые плоды
|
| лимонная
| HOOCCH2(OH)COOHCH2COOH
| 
| 64,04
| цитрусовые плоды и ягоды
|
| винная
| HOOCCH(OH)CH(OH)COOH
|
| 75,04
| виноград
|
Дополнительная литература
1. Коренман Я.И., Лисицкая Р.П. Практикум по аналитической химии. Анализ пищевых продуктов. – Воронеж: Воронеж. гос. технол. акад., 2002.
Введение
Лабораторная работа выполняется на современном приборе – вольтамперометрическом анализаторе «Экотест-ВА». Установка состоит из электрохимической ячейки, измерительного преобразователя (ИП) и IBM-совместимого персонального компьютера с установленным пакетом программного обеспечения, с помощью которого осуществляется управление процессом анализа, обработка и хранение данных.
Определение тяжелых металлов – цинка, кадмия и свинца – выполняется с рабочим углеситалловым электродом методом переменнотоковой инверсионной вольтамперометрии. Для анализа сильно загрязненных образцов воды и пищевых продуктов необходима предварительная минерализация проб. Анализ модельных растворов и питьевой воды такой пробоподготовки, как правило, не требует, но при низкой концентрации тяжелых металлов в пробе рекомендуется концентрирование раствора.
Экспериментальная часть
Оборудование и посуда: анализатор вольтамперометрический «Экотест-ВА»; компьютер; электрохимическая ячейка; рабочий углеситалловый электрод, хлорсеребряный электрод сравнения, вспомогательный платиновый электрод; магнитная мешалка; фарфоровые чашки.
Реактивы: стандартные растворы солей цинка, кадмия и свинца – по 10 мкг/мл в пересчете на соответствующий катион; раствор HCl 1 М; фоновый электролит (в 1 л р-ра: 50 мл 1 М НСl, 10 мл 0,01 М Hg(NO3)2); концентрированная HNO3.
Внимание! Для приготовления растворов и ополаскивания посуды вместо дистиллированной воды используется бидистиллированная!
Подготовка установки к работе:
1. Перед работой корпус ИП необходимо заземлить.
2. Поверхность рабочего углеситаллового электрода очистите ватой, смоченной спиртом, и ополосните бидистиллятом.
3. Подключите подготовленные к работе электроды к соответствующим разъемам на передней панели ИП: рабочий углеситалловый, хлорсеребряный электрод сравнения и вспомогательный платиновый. Закрепите электроды в штативе и опустите в ячейку с 25 мл фонового электролита и якорьком магнитной мешалки.
4. К разъемам на задней панели ИП подключите мешалку (разъем «УПР»), блок питания и соединительный кабель для подключения к СОМ-порту компьютера.
5. Включите ИП тумблером «Сеть», при этом на передней панели загорится индикаторный светодиод.
6. Включите компьютер и запустите программу: «n_va2010». Появится главное окно управления (рис. 2).
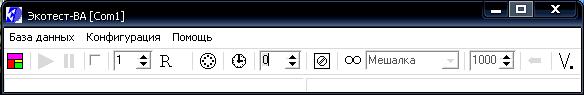
Рис. 2. Главное окно управления программы «n_va2010».
7. Активируйте связь с ИП, нажав кнопку «Window» на панели управления анализатором. При правильном выборе и подключении СОМ-порта светодиод на передней панели ИП будет «моргать» несколько секунд, а в программе появится индикация установившейся связи с прибором (рядом с кнопкой «R – сброс ВА»). При отсутствии информационного обмена между ИП и ПК на экране монитора появится сообщение об ошибке.
8. Создайте и сохраните новую базу данных: во вкладке «База данных» выберите пункт «Новая». Появятся три окна: «Параметры развертки», «Вычисление» и «График». Во вкладке «База данных» также выберите и нажмите на пункт «Запоминать изменения».
Выполнение измерений:
1. В окне «Параметры развертки» (рис. 3) установите следующие значения:
Очистка: 0 мВ, 60 с.
Подготовка 1 и подготовка 2: 0 с.
Накопление: –1300 мВ, 60 с.
Мешалка: активна, время успокоения раствора – 10. Скорость вращения мешалки выставляется поворотом ручки на ее корпусе.
Диапазон тока: 2мА/200 мкА.
Развертка: скорость – 50 мВ/с, начало: –1300 мВ, конец: –200 мВ.
Схема соединения: 3х электродная.
Тип измерения: Фон.
Модуляция: активна, 30 мВ/25Гц (амплитуда переменного напряжения).
Комментарий: Zn Cd Pb
В последующих экспериментах необходимо менять «Тип измерения» и «Комментарий».
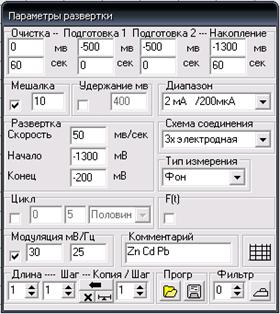
Рис. 3. Окно «Параметры развертки»
2. В окне «Вычисление» (рис. 4) проверьте, какие определяемые элементы отображаются (должны быть Zn, Cd, Pb). В случае несоответствия удалите (Ó) и/ или добавьте (Å) необходимый элемент в верхней строчке окна.

Рис. 4. Окно «Вычисление».
3. В окне «График» (рис. 5) установите масштаб по оси Х от -1300 до -200, по оси Y от 30 до 200.
4. Задайте число повторов (2 – 3) в главном окне управления анализатором (рядом с «R») и нажмите «Старт». Начнется измерение – очистка рабочей поверхности электрода, накопление, развертка потенциала. По окончании измерений прозвучит звуковой сигнал, и в окне «График» появятся вольтамперограммы для фонового раствора. Условия измерений аналогичны для всех опытов.

Рис. 5. Окно «График».
Обработка вольтамперограмм:
1. Для вычисления площадей (высот) пиков элементов в окне «График» для выделенной вольтамперограммы нажмите «Установить границы» ( ). Внизу окна «График» появится соответствующий инструмент. Для каждого определяемого катиона необходимо выбрать начало и конец пика, как показано на рис. 6.
). Внизу окна «График» появится соответствующий инструмент. Для каждого определяемого катиона необходимо выбрать начало и конец пика, как показано на рис. 6.

Рис. 6. Установка границ пиков металлов на вольтамперограмме.
После выбора границ необходимо нажать кнопку «Запомнить и выйти».
2. В таблице «Вычисление» будет отображаться результат: площади пиков соответствующих элементов или высоты (переключение между ними – кнопками «S» или «Н»).
3. При работе с сохраненной ранее базой данных необходимо выбрать пункт «Чтение из базы». Появится окно со всеми записями (рис. 7). Для проведения вычислений площадей (высот) пиков выбираем нужную запись и, кликнув на нее правой кнопкой мыши, выбираем «Добавить в таблицу». Добавленную вольтамперограмму можно обрабатывать.

Рис. 7. Окно «База данных».
Ход работы
Опыт 1. Определение цинка, кадмия и свинца в модельном растворе
1. Получите вольтамперограммы фонового раствора (3 – 4 раза).
2. Приготовьте раствор с известными концентрациями катионов цинка, кадмия и свинца – по 0,1 – 0,2 мкг/мл. Для этого в мерную колбу на 25,0 мл поместите с помощью мерных градуированных пипеток рассчитанные объемы исходных стандартных растворов солей металлов, доведите раствор до метки фоновым электролитом.
3. Поместите приготовленный раствор в чистую сухую ячейку и проведите измерения. Повторить запись вольтамперограммы 2-3 раза.
4. Не извлекая электроды из раствора, внесите в ячейку мерной пипеткой добавки стандартных растворов солей металлов. Объем вводимой добавки надо подобрать так, чтобы концентрации катионов металлов в результате ввода добавки возросли примерно вдвое.
5. Последовательно получите несколько вольтамперограмм раствора с добавкой.
6. Введите в раствор новую добавку (такие же объемы стандартных растворов солей металлов, как в случае первой добавки по п. 4) и повторите измерения.
Обработка результатов
Проведите вычисление площадей пиков по всем полученным вольтамперограммам, рассчитайте концентрации катионов металлов в модельной смеси и сопоставьте с действительными значениями.
Опыт 2. Анализ питьевой воды на содержание цинка, кадмия и свинца
1. Пробу подготовленного к испытанию образца воды объемом 100 мл поместите в выпарительную фарфоровую чашку, добавьте 1-2 мл концентрированной азотной кислоты. Содержимое чашки выпарите до «влажных солей». Параллельно в другой чашке подготовьте таким же образом «холостую» пробу, используя вместо пробы бидистиллированную воду.
2. После обработки азотной кислотой в чашку добавьте 1 мл 1 М HCl и 5 мл фонового электролита. Полученный раствор количественно перенесите в мерную колбу на 25,0 мл, чашку ополосните несколько раз небольшими порциями фонового электролита. Доведите раствор до метки фоновым электролитом.
3. Получите вольтамперограммы «холостой» пробы и пробы воды. В зависимости от того, пики каких катионов имеются на вольтамперограмме анализируемой воды, введите добавки стандартных растворов соответствующих солей и получите вольтамперограммы пробы с добавкой.
Обработка результатов
Проведите обработку данных, рассчитайте концентрации цинка, кадмия и свинца в анализируемой воде. При необходимости учтите «холостую» пробу. Рассч