ВВЕДЕНИЕ
Нефтехимический синтез – производство химических продуктов на основе нефтяного углеводородного сырья – получил огромное развитие во второй половине XX века. Использование нефтяного сырья привело к большому прогрессу в химической промышленности, и особенно в производстве полимеров, на выработку которых расходуется основная масса углеводородного сырья. В промышленности основного органического и нефтехимического синтеза сосредоточены важнейшие производства мономеров, исходных и вспомогательных продуктов, различных добавок для полимерных материалов. При этом технологии производства этих соединений во многом отличаются от технологий производства высокомолекулярных соединений. Будущим инженерам и руководителям производств синтетических каучуков и других полимеров необходимо знание процессов нефтехимического синтеза еще и потому, что в последнее время наблюдается тенденция размещения в рамках одного предприятия производств мономеров и полимеров на их основе.
В настоящих методических указаниях дается основа для обучения экспериментальным методам нефтехимического синтеза. Включены работы, охватывающие наиболее распространенные процессы – дегидрирование, конденсация, алкилирование, эпоксидирование, окислительные методы. В каждой работе приведены краткие теоретические основы способа, описание экспериментальной установки, порядок выполнения, методики контроля и анализа образующихся продуктов, расчет показателей процесса.
Основной целью практикума является освоение студентами способов и приемов, используемых в нефтехимическом синтезе, методов управления и контроля процессами, расчета материальных балансов и показателей процесса.
Лабораторная работа 1
Описание установки
Установка собрана на основе препаративного хроматографа ПАХВ-05. Принципиальная схема установки приведена на рис. 1.
 Рис. 1. Принципиальная схема микрокаталитической установки дегидрирования в импульсном режиме:
Рис. 1. Принципиальная схема микрокаталитической установки дегидрирования в импульсном режиме:
1,3 - вентили тонкой регулировки; 2 - манометр образцовый; 4 - автоматический объемный дозатор; 5 - шестиходовой газовый дозирующий кран; 6 - испарители; 7 - реактор; 8 -хроматографические колонки; 9 - детектор по теплопроводности (ДТП)
Гелий из баллона подается в систему вентилей тонкой регулировки 1,3. Общее давление газа-носителя на входе фиксируется манометром 2. При помощи вентиля 3 гелий подается в дозатор, который служит для дозирования жидкого сырья. Вентиль 3’’ подает гелий на сравнительную линию хроматографа, которая состоит из испарителя 6’ и хроматографических колонок 8’. Из колонки 8’ гелий попадает в сравнительную камеру ДТП 9. Вентиль 3’ регулирует поток гелия в рабочую линию хроматографа, которая состоит из образцового манометра 2’, шестиходового дозирующего крана 5, испарителя 6, реактора 7 и хроматографической колонки 8. Из колонки 8 гелий попадает в рабочую камеру ДТП 9. Реактор 7 представляет собой трубку из титана длиной 216 мм и внутренним диаметром 5 мм. В реактор загружается кварцевая насадка фракции 0,16-0,5 мм, которая удерживается асбестовыми пробками. Затем загружается 0,4 г катализатора фракции 0,2-0,5 мм (слой катализатора находится в плато печи). Реактор обогревается трубчатой электрической печью, температура которой регулируется ЛАТРом и фиксируется термопарой, которая помещена между реактором и печью. Шестиходовой кран 5 служит для дозирования импульса сырья (бутана).
Хроматографические колонки 8 наполнены диамитовым кирпичом (фракция 0,15-0,25 мм), пропитанным 15%-ным бутиратом триэтиленгликоля. Длина колонок 7 метров.
Выполнение работы
1. Загрузить катализатор. Открутив гайки, снять реактор, предварительно вынуть термопару. Загрузить в реактор 0,4 г катализатора ИМ-2201 фракции 0,2-0,5 мм. Подсоединить реактор, подтянуть гайки и вставить термопару до середины печи.
2. Открыть газ-носитель (гелий) и установить необходимое давление на манометрах (2) и (2') (~ 43-45 делений).
3. Проверить установку на герметичность: разница показаний на манометрах (2) и (2') не должна превышать 3-4 деления. Если разница в показаниях манометров превышает 3-4 деления, значит, гайки на реакторе надо затянуть потуже.
4. Включить обогрев реактора. Для этого включить в сеть ЛАТРы и милливольтметр. Положение стрелки милливольтметра на 150 В, затем 170 В и через 30 минут 180-190 В, что соответствует 630-650 °С.
Температура в термостате должна быть 35-45 °С - контролируется по термометру. Общее время выхода на режим 1-1,5 часа.
5. Провести регенерацию катализатора. После установления температуры 630-650 °С дозировочный кран 5 установить в положение 2 (сброс на воздух), отсоединить кран от баллона с бутаном (снять резиновую трубку), продуть грушей и перевести в положение 1. В положении 1 подать грушей 20-25 импульсов воздуха с интервалом 20-30 секунд.
6. Провести дегидрирование бутана. Подсоединить кран 5 к баллону с бутаном и перевести его в положение 2. Охладить реактор до 570 °С (убавить напряжение на ЛАТРе до 165-170 В). Одновременно, не дожидаясь охлаждения, включить тумблер 2 на блоке детектора и самописец. По достижении температуры реактора 570 °С и температуры термостата колонок 35-40 °С установить точный расход газа носителя по МПР. Расход гелия -50 мл в минуту (10 мл за 11-12 секунд). Ручками "грубо" и "точно" установить нулевую линию самописца при положении чувствительности 3 (ручка 13).
Открыть баллон с бутаном и по пробулькиванию пузырьков газа в воде контролировать подачу бутана в дозирующий кран 5 (положение 2).
Включить ленту самописца и перевести кран 5 в положение 1 (выдержка ~1 минута). Пока выписывается хроматограмма, баллон с бутаном можно закрыть. При выписывании первых пяти пиков ручка 13 находится в положении 3, после пятого пика ручка 13 переводится на 6 масштаб, в этом положении выписывается один пик (бутан), затем переключаем на 5 масштаб - выписывается еще 4 пика, и ручка 13 снова ставится в положение 3. После выхода на нуль вводим новый импульс бутана. Всего вводится 10 импульсов.
7. Отключить обогрев реактора. После охлаждения его для определения состава исходного углеводородного сырья провести хроматографирование бутан-содержащего газа в описанных выше условиях.
8. Выключитъ тумблеры КСП и тумблер 11 на блоке детектора. Отключить щиток. Закрыть баллон с гелием.
Техника безопасности
Лабораторная работа на установке дегидрирования в импульсном режиме относится к взрыво-, пожаро- и электроопасным, поскольку используемое сырье – бутан и получаемые продукты – бутадиен и бутилены – являются легковоспламеняющимися веществами, а пары их с воздухом могут образовывать смеси, взрывающиеся при соприкосновении с источником воспламенения (искра, пламя, нагретая поверхность).
Работу на установке дегидрирования следует проводить только на исправном оборудовании в строгом соответствии с методическими указаниями при полной герметичности системы.
Не допускать утечки паров и газа!
Работающую установку не оставлять без наблюдения, во время работы необходимо осуществлять контроль за работой электрической схемы. Все электроприборы должны иметь защитное заземление.
По окончании работ установка должна быть отключена от источников питания.
В случае аварийного состояния оборудования немедленно отключить установку от сети и прекратить подачу сырья. В случае возникновения пожара немедленно эвакуировать баллон с бутаном из помещения, после чего принять меры по тушению пожара с помощью асбестового одеяла, песка и углекислотного огнетушителя.
Лабораторная работа 2
Описание установки
Дегидрирование этилбензола в стирол проводят на установке, схема которой представлена на рис. 2.

Рис. 2. Установка для дегидрирования этилбензола:
1 - электронагревательная печь; 2 - реактор; 3 - бюретка для воды; 7,4 - микронасосы; 5 - испарители; 6 - бюретка для сырья; 8 - холодильник; 9 - приёмник; 10 -термопара; 11 - потенциометр; 12 - охлаждающая баня со льдом
Установка состоит из системы дозирования и испарения сырья, реакторного блока и узла конденсации. Реактор 2 изготовлен из нержавеющей стали, представляет собой трубку диаметром 22x2 мм, нижняя часть которой переходит в узкий канал диаметром 10 мм. В верхней части реактора имеются штуцера для ввода перегретого пара, сырья и воздуха. В реактор вставляют решетку, которая обеспечивает нахождение катализатора в плато печи в зоне максимальной и относительно постоянной температуры. Решетка изготовлена из нержавеющей стали. В центр реактора вставлен карман, в котором размещена термопара ХА (хромель-алюмелевая).
Для определения плато печи проводят предварительные эксперименты. Для этого печь разогревают до 600 °С и дают выдержку 2 часа. После чего промеряют температуру через 1 см по высоте печи при подаче водяного пара в количестве 70-80 мл/ч. Плато печи составляет 5 см.
Испаритель 5 представляет собой трубку диаметром 40x2 мм, длиной 250 мм, изготовленную из стали Х18Н9Т. Испаритель помещен в электронагревательную печь. Обогрев печи реализуется с помощью ЛАТРа. С внешней стороны испаритель изолирован асбестом. Микронасосы 4,7 предназначены для подачи сырья и воды в реактор. Производительность насоса 0-240 мл в час.
Выполнение работы
Для дегидрирования этилбензола используется промышленный катализатор К-24 фракции 2÷3 мм. Объём загрузки катализатора составляет 10 см3. Реактор заполняют катализатором и инертной насадкой (кварц), как показано на рис.2. Свежезагруженный катализатор перед контактированием подвергается активизации паровоздушной смесью. Для этого катализатор нагревается в токе воздуха со скоростью 200 °С в час до 400 °С. Подача воздуха поддерживается 700 л/л катализатора в час (0,28 л/мин). Начиная с 400 °С подаётся вода со скоростью в 2 раза меньшей, чем при контактировании. При температуре 600 °С катализатор выдерживается 1 час в токе паровоздушной смеси, после чего проводится дегидрирование.
Исходный зтилбензол и вода заливаются в бюретки, и отмечается их уровень. При достижении заданной температуры в течение 10 мин подаётся вода из бюретки с помощью микронасосов, а затем этил бензол.
Водяной пар вводится в реактор в строго дозированном количестве. Для этого дистиллированная вода из бюретки 3 насосом 4 подаётся в испаритель 5, испаряется и образующийся пар перегревается. Далее перегретый пар поступает в реактор. Температуру контактирования измеряют термопарой, работающей в комплекте с потенциометром ЭПВ 2-11 А.
Смесь этилбензола и водяного пара проходит по реактору до слоя катализатора, нагревается до температуры реакции и поступает на катализатор. На неподвижном слое катализатора проходит эндотермическая реакция дегидрирования. Смесь контактного газа с водяным варом, выходящая из реактора 2, охлаждается, конденсируется в холодильнике 8 и поступает в приемник 9. Несконденсировавшаяся часть контактного газа выбрасывается через воздушку. Отбор пробы реакционной массы для анализа начинают после 15 мин. стабильной работы установки на заданном режиме. Органический слой (печное масло) отделяют от водного в делительной воронке, взвешивают его, сушат над безводным хлоридом кальция и анализируют на хроматографе, определяя количественный и качественный составы. Хроматографическая колонка длиной 1,0 м заполнена 15%-ным гексакис-β-цианэтоксигексаном на кирпиче фракции 0,16-0,25. Оптимальные условия работы хроматографа: Т колонки- 100 °С, Т испарителя - 200 °С, скорость диаграммной ленты - 600-720 мм/ч.
Процесс дегидрирования проводится при следующих условиях:
Т = 580 °С, 600 °С, 620 °С (указывается преподавателем).
Массовое соотношение ЭБ: Н2О =1:3.
Скорость подачи ЭБ - 0,5 ч-1, 1,0 ч-1 (0,2 мл/мин, 0,4 мл/мин).
После каждого опыта при температуре контактирования проводится регенерация катализатора пропусканием паровоздушной смеси. Для этого сначала подаётся вода, скорость подачи которой при регенерации равна скорости воды при контактировании, затем подаётся воздух. Скорость подачи воздуха регулируется таким образом, чтобы температура в реакторе не превышала 650 °С. Регенерацию ведут в течение 30 мин, затем прекращают подавать воздух, воду продолжают подавать в течение 30 мин, после чего можно вновь проводить контактирование. Результаты опытов записывают в табл. 2.
Таблица 2. Результататы экспериментов
| №
п/п
| Темпе-ратура, °С
| Объемная скорость подачи ЭБ, ч -1
| Состав жидких продуктов реакции, %мас.
| Конверсия, %
| Селективность, %
|
|
|
|
| бензол
| толуол
| ЭБ
| стирол
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

Образец хроматограммы продуктов дегидрирования стирола
Техника безопасности
Стирол имеет молекулярную массу 104,14, температуру кипения 145,2 °С, плотность 0,9060 г/см3.
Стирол представляет собой бесцветную прозрачную сильно преломляющую свет жидкость с характерным запахом. Смешивается во всех отношениях с метиловым и этиловым спиртами, ацетоном, диэтиловым эфиром, бензолом, толуолом, четырёххлористым углеродом. Он является хорошим растворителем для многих соединений. В воде растворяется плохо.
Стирол характеризуется высокой реакционной способностью. На воздухе легко окисляется с образованием альдегидов и кетонов, придающих ему неприятный запах. Стирол является довольно летучим и легко воспламеняющимся веществом с низкими пределами взрываемости смеси его паров с воздухом.
Пределы взрываемости с воздухом - 1,1-5,2 % об.
Токсичен. Жидкий стирол раздражает кожные покровы и при длительном воздействии вызывает воспалительные процессы. Пары стирола раздражают слизистые оболочки глаза и носа, интенсивность раздражения увеличивается с увеличением концентрации.
Предельно допустимая концентрация стирола в воздухе производственных помещений 5 мг/м3. Все работы со стиролом проводить обязательно в вытяжном шкафу.
Лабораторная работа 3
Техника безопасности
4,4-диметилдиоксан-1,3 - бесцветная, прозрачная, однородная жидкость. Температура кипения -133,4 °С; плотность- 0,963 г/см3.
ДМД действует на организм человека наркотически, возбуждает нервную систему. Признаки отравления человека: головная боль, головокружение, общая слабость, чувство опьянения, бледность, частый пульс, иногда рвота, а в более тяжелых случаях - потеря сознания. При первых признаках отравления пострадавшего немедленно вывести на свежий воздух.
Формальдегид (муравьиный альдегид, метаналь) НСНО – раздражающий газ с резким запахом. Хорошо растворим в воде, спиртах, умеренно – в бензоле, эфире, хлороформе. Формалин (формоль) – водный раствор формальдегида (обычно 37-40 %-ный), мутнеет при хранении из-за выпадения белого осадка параформальдегида. Растворы выделяют газообразный формальдегид даже при комнатной температуре. Газообразный формальдегид горит, с воздухом или кислородом образует взрывоопасные смеси. Концентрационные пределы взрываемости – 7,0 -7,2 %. Формальдегид вызывает дегенеративные процессы в паренхиматозных органах, сенсибилизирует кожу, обладает мутагенными свойствами, оказывает легкое раздражающее действие на слизистые оболочки глаз и дыхательных путей. ПДК 0,05 мг/м3. При попадании на кожу – немедленно промыть водой, лучше 5 % -ным раствором нашатырного спирта. При отравлении через рот – незамедлительное промывание желудка 3 % -ным раствором карбоната аммония.
Альфаметилстирол (изопропенилбензол) С6Н5С(СН3)=СН2 – подвижная бесцветная жидкость с резким специфическим запахом. Альфаметилстирол смешивается во всех соотношениях с ацетоном, четыреххлористым углеродом, бензолом, н-гептаном и этанолом, растворимость альфаметилстирола в воде – 0,01 % (по объему). Твсп. =58 °С. Альфаметилстирол оказывает слабое раздражающее действие на слизистые оболочки глаз, носа и горла при концентрации 0,05 мг/л в течение 10-30 секунд, хорошо всасывается через кожу.
Лабораторная работа 4
Выполнение работы
Разложение ГПИПБ проводят на установке, изображенной на рис. 4. Перед началом опыта проверяют правильность сборки, герметичность всех соединений, работу термостата и механической мешалки. В реактор загружают 25 мл изопропилбензола, 0,15 мл концентрированной серной кислоты. Включают подачу воды в обратный холодильник, механическую мешалку и термостат (обогрев бани и электрореле). Процесс проводят при температуре 50 °С, которую поддерживают с точностью до 0,5 °С. Внимание!!! Строго следить за поддержанием точной температуры во избежание ускорения разложения ГПИПБ, способного привести к взрыву.
Внимание!!! Строго соблюдать порядок ввода реагентов в реакционную колбу.
По достижении заданной температуры и при энергичном перемешивании к раствору серной кислоты в изопропилбензоле из капельной воронки осторожно прибавляют 50 мл 15 %-ного раствора ГПИПБ. По окончании прибавления ГПИПБ каждые 30 минут отбирают по две пробы реакционной массы (по 1 мл) и анализируют на содержание в ней гидропероксида. Концентрацию ГПИПБ в пробах определяют иодометрически.
Если по истечении 1,5 часов разложение ГПИИПБ не завершится, то к реакционной массе добавляют еще 0,25 мл концентрированной серной кислоты и продолжают опыт до полного разложения ГПИПБ.
Результаты анализов и вычислений записывают в табл. 4.
По окончании опыта содержимое реактора охлаждают до комнатной температуры, определяют объем, массу и анализируют на содержание ацетона. Все операции проводят таким образом, чтобы свести к минимуму потери ацетона за счет улетучивания.

Рис.4. Установка для разложения гидропероксида изопропилбензола
Таблица 4. Результаты эксперимента
| № пробы
| Время отбора пробы, мин
| Масса пробы, г
| Объем 0,1н раствора тиосульфата натрия, пошедший на титрование пробы, мл
| Объем 0,1н раствора тиосульфата натрия, пошедший на титрование холостого опыта, мл
| Концентрация ГПИПБ, % мас.
|
| | | | | | |
Составляют материальный баланс опыта (табл.5).
Таблица 5. Материальный баланс опыта
| Наименование компонентов
| Приход, г
| Получено, г
|
| ГПИПБ
|
|
|
| Фенол
| -
|
|
| Ацетон
| -
|
|
Селективность процесса, % мол.
по фенолу-
по ацетону-
 Массу ацетона, образовавшегося в результате разложения ГПИПБ, рассчитывают по формуле:
Массу ацетона, образовавшегося в результате разложения ГПИПБ, рассчитывают по формуле:
i
где: Ga -масса ацетона, г; Gpm - масса реакционной массы, выгруженной из реактора, г; Ха- массовая доля ацетона в продуктах реакции; Gi - масса i пробы, г; n- число проб.
Массу полученного фенола определяют как разницу между количеством ГПИПБ, взятого в реакцию, и количеством полученного ацетона.
Обсуждают данные, полученные в работе, и формулируют свои выводы по работе.
Анализ реакционной массы на содержание ацетона
В две конические колбы на 250 мл с притертыми пробками наливают 25 мл 0,5 н раствора гидроксиламина солянокислого, приливают по 5 капель индикатора бромфенолового синего и нейтрализуют 0,1 н раствором гидроксида натрия до появления синей окраски раствора. Затем добавляют пипеткой по 1 мл реакционной массы, закрывают пробки, встряхивают в течение 20 минут и оттитровывают выделившуюся соляную кислоту 0,1 н раствором гидроксида натрия до появления синей окраски.
Параллельно определяют количество 0,1 н раствора гидроксида натрия, необходимое для нейтрализации серной кислоты, содержащейся в 1 мл реакционной массы (холостой опыт). Для этого в коническую колбу на 250 мл с притертой пробкой наливают 25 мл дистиллированной воды, приливают 5 капель индикатора бромфенолового синего и 1 мл реакционной массы и при встряхивании нейтрализуют 0,1 н раствором гидроксида натрия до появления синей окраски.
Глубина окраски раствора при всех титрованиях должна быть одинакова.
Содержание ацетона (X, % маc.) в реакционной массе вычисляют по формуле:
X=[a-(b+c)]K·0,00695/V,
где: а - объем 0,1 н раствора NaOH, пошедший на титрование пробы с ацетоном, мл; b - объем 0,1 н раствора NаОН, пошедший на титрование солянокислого гидроксиламина, мл; с - объем 0,1 н раствора NаОН, пошедший на титрование 1 мл реакционной массы, мл; К - поправка на 0,1 н раствор NаОН; 0,00695 - число граммов ацетона, соответствующее 1 мл 0,1 н раствора NаОН; V - объем реакционной массы, взятый на анализ, мл.
Оказание первой помощи
Немедленно удалить продукты, попавшие на кожу, тампоном, смоченным спиртом, затем пораженный участок тела обильно промыть водой с мылом. При попадании в глаза - немедленно провести обильное и длительное (10-15 мин.) промывание проточной водой, затем срочно обратиться к врачу. Ни в коем случае не промывать глаза маслом или маслосодержащими жидкостями. Это усиливает поражающее действие.
При ингаляционном отравлении необходимо обеспечить пострадавшему покой, свежий воздух. При необходимости - кислород, сердечные средства, обязательно обратиться к врачу.
Лабораторная работа 5
Эпоксидирование a-олефинов С8 – С10 гидропероксидом этилбензола
Реакция эпоксидирования олефинов органическими гидропероксидами позволяет одновременно получить два целевых продукта - оксид олефина и спирт. Спирт обычно затем дегидратируют в соответствующий олефин.
В промышленности процесс эпоксидирования олефинов гидропероксидами используют для совместного получения оксида пропилена и стирола (Халкон-процесс) или изобутилена и других продуктов.
Процессы обычно состоят из трех стадий:
1) окисление соответствующего углеводорода в гидропероксид:

2) эпоксидирование олефина полученным раствором гидропероксида:
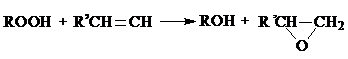
3) дегидратация полученного спирта в олефин:

Катализаторами эпоксидирования олефинов гидропероксидами являются соли и комплексы молибдена, вольфрама, ванадия и др. переходных металлов, растворимых в реакционной массе, среди которых наиболее активными и селективными являются комплексы молибдена.
Схема процесса может быть представлена следующим образом:

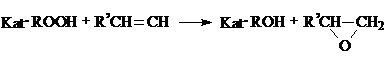

В промышленности процесс эпоксидирования обычно осуществляют в растворе того же углеводорода, из которого получен гидропероксид, при температуре 80-100 °С, давлении до 7 МПа, мольном отношении гидропероксцда к олефину, равном 1: (2 - 5), и концентрации катализатора 0,001-0,005 моль на моль гидропероксида. В зависимости от природы исходных реагентов и условий процесса время реакции может составлять от 0,3 до 2 часов.
Цель работы: изучение влияния концентрации катализатора (или гидропероксида) на скорости расходования гидропероксида, образования оксида a-олефинов С8 – С10 и на селективность процесса по гидропероксиду этилбензола.
Реактивы:
1)a-олефины С8 – С10- средняя ММ - 126, Ткип= 121,27-170,6 °С, d420 = 0,715-0,730;
2) гидропероксид этилбензола, 30 %-ный раствор - ММ = 138, Ткип= 136,2 °С, d420= 1,074; nd20 = 1,527;
3) этилбензол - ММ-106,2, Ткип = 136 °С, d420 = 0,867, nd20 =1,4959;
4) ацетилацетонат молибденила (АсАс)2МоО2 – ММ=326, либо комплексный молибденовый катализатор – КМК (содержание растворимого молибдена равно 0,3-0,5 % мас.); АМ Mo=96;
5) ледяная уксусная кислота - ММ=60, Ткип = 118,1 °С, d420 = 1,0492, nd20 = 1,3730;
6) 50 %-ный водный раствор йодистого калия;
7) 0,1н раствор тиосульфата натрия;
8) 5 %-ный водный раствор крахмала;
9) 0,1н раствор гидроксида натрия;
10) 0,2н раствор соляной кислоты в ацетоне;
11) 1 %-ный спиртовый раствор фенолфталеина;
12) карбонат натрия.
Посуда и оборудование: четырехгорлый реактор из термостойкого стекла; обратный холодильник; термометр на 150°С; механическая мешалка с электромотором и ЛАТРом; водяная баня, снабженная контактным термометром с электрореле; микропипетки на 1 мл - 3 штуки; мерный цилиндр на 30-50 мл, мерный цилиндр на 10 мл; конические колбы на 250 мл - 3 штуки; конические колбы на 100 мл - 3 штуки; бюретки для титрования - 2 штуки.
Выполнение работы
a-олефины С8 – С10 эпоксидируют на установке, изображенной на рис. 6. Перед началом опытов проверяют правильность сборки, герметичность соединений установки и надежность работы механической мешалки и термостатирующей установки.
Для изучения влияния концентрации катализатора на скорость эпоксидирования и селективность процесса проводят серию опытов при постоянной температуре реакционной массы 60 ± 0,5 °С, концентрации a-олефинов С8 – С10 2 моль/л (17,64 г), концентрации гидропероксида этилбензола (ГПЭБ) 0,5 моль/л (4,83 г или 34,5 г 14 % раствора) и общем объеме ис-ходных реагентов, равном 70 мл при трех различных концентрациях молибденового катализатора: 1, 3, 6 ммоль/л.
| Рис 5. Установка для эпоксидирования a-олефинов С8 – С10
|

Прежде чем приступить к работе, исходя из заданных начальных концентраций катализатора, рассчитывают объемы исходных реагентов и катализатора. Результаты вычислений записывают в табл. 6.
Таблица 6. Объемы исходных реагентов и катализатора
| № опыта
| Конц-ция ката-лизатора, ммоль/л
| Объем раствора ката-лизатора, мл
| Конц- ция раствора ГПЭБ, моль/л
| Объем раствора ГПЭБ, мл
| Объем
a-олефинов С8 – С10, мл
| Объем этил-бензола, мл
|
|
|
|
| 0,5
|
|
|
|
|
|
|
| 0,5
|
|
|
|
|
|
|
| 0,5
|
|
|
|
Внимание!!! Строго следить за поддержанием точной температуры во избежание ускорения разложения ГПЭБ, способного привести к взрыву.
В сухой, чистый реактор наливают рассчитанные объемы растворов гидропероксида этилбензола, a-олефинов С8 – С10 и этилбензола. Включают мешалку, термостатирующую установку, подачу воды в обратный холодильник. По достижении в реакторе заданной температуры к реакционной массе добавляют рассчитанное количество раствора молибденового катализатора. Момент добавления катализатора принимают за начало процесса.
В ходе опыта отбирают по две параллельные пробы реакционной смеси для анализа на содержание ГПЭБ и оксида. Пробы отбирают калиброванной пипеткой на 1 мл каждые 5 минут в течение первых 20 минут, а затем каждые 20 минут Продолжительность опыта в зависимости от концентрации катализатора составляет от 1 часа до 2,5 часа. По окончании опыта реакционную массу охлаждают и остатки гидропероксида разлагают, прибавляя небольшими порциями карбонат натрия, не допуская разогрева реакционной массы.
На основании полученных данных строят зависимости в координатах концентрация ГПЭБ либо оксида a-олефинов С8 – С10 - время при различных начальных концентрациях катализатора. Результаты анализов и вычислений записывают в табл. 7.
Таблица 7. Результаты анализов и вычислений
| Время отбора проб, мин.
| Объем 0,1н NaOH, пошедший на титрование, мл
| Концентра-ция оксида, % мас.
| Объем 0,1н Na2S2O3, пошедший на титрование, мл
| Концентрация ГПЭБ, % мас.
| Селективность по ГПЭБ, %
|
|
|
|
|
|
|
|
Лабораторная работа 6
Выполнение работы
Алкилирование фенола тримерами пропилена или a-олефинами С8 – С10 на катионите проводят на установке, изображенной на рис.6.

| Рис.6. Установка для алкилирования фенола
| |
Перед началом опытов проверяют правильность сборки, герметичность соединений установки и надежность работы механической мешалки и термостатирующей установки. Термостатирующую установку настраивают на поддержание в реакторе температуры 80 °С.
В колбу-реактор загружают 33 г фенола и 30 г катионита. Катионит предварительно переводят в водородную форму и обезвоживают, как описано ниже.
Подают воду в обратный холодильник и включают обогрев реактора. Механическую мешалку включают после расплавления фенола. По достижении температуры реакционной массы 80 °С начинают подачу тримеров пропилена или a-олефинов С8 – С10 в количестве 40 г. После окончания подачи алкилирующего агента реакционную массу выдерживают еще 90 минут, при перемешивании повышая температуру до 120 °С, охлаждают до 60 °С и быстро фильтруют на воронке Бюхнера при отсасывании водоструйным насосом.
Фильтрат переливают в предварительно взвешенную колбу Кляйзена, вновь взвешивают и перегоняют при атмосферном давлении или в вакууме с использованием воздушного холодильника.
При перегонке выделяют фракции, кипящие в различном интервале температур, и определяют их массу в граммах (табл.8).
Таблица 8. Температуры кипения различных фракций
| Фракция
| Ткип при атм. давлении, °С
| Ткип при 10 мм рт. ст,°С
|
| Фенол (в основном)
| до 185
| до 90
|
| Промежуточная
| 185-
| 90-140
|
| н-Нонилфенол
|
| 140-170
|
| Остаток
|
| >170
|
Катионит из воронки Бюхнера переносят в чистую колбу, закрывают пробкой и хранят до повторного использования.
На основе полученных данных составляют материальный баланс опыта:
Подано на реакцию, г:
фенола -
тримеров пропилена –
Получено реакционной массы, г-
Выделено из реакционной массы, г:
фенола –
п-нонилфенола -
Выход п-нонилфенола по фенолу, % мас. –
Потери:
г
% мас.
Обсуждают результаты опыта и формулируют выводы по проделанной работе.
Подготовка катионита для алкилирования
Катионит, используемый в качестве катализатора в процессе алкилирования, должен находиться в кислой (водородной) форме. Для перевода катионита в кислую форму его обрабатывают при комнатной температуре 10%-ным раствором соляной или серной кислоты на установке, изображенной на рис. 7.


На дно первой делительной воронки емкостью 200 мл кладут тампон из стеклянной ваты, загружают 50 г катионита. Воронку закрепляют на штативе и при закрытом кране катионит заливают раствором кислоты так, чтобы уровень кислоты над катионитом составлял 15-20 мм. Во вторую делительную воронку с закрытым краном емкостью 500-700 мл заливают 500 мл кислоты и устанавливают ее над первой делительной воронкой, закрепив на штативе. Затем открывают краны обеих воронок так, чтобы обеспечить прохождение кислоты через слой катализатора со скоростью 1 мл/мин. Обработку проводят до тех пор, пока концентрация кислоты, выходящей из слоя катализатора, не будет равна концентрации кислоты, подаваемой на слой катализатора. Концентрацию кислоты определяют титрованием. Первую проверку проводят через 6 часов, а затем каждый час. После этого кислоту из обеих воронок сливают, и катионит промывают в делительной воронке дистиллированной водой до нейтральной реакции по метиловому оранжевому.
Затем катионит сушат сначала между слоями фильтровальной бумаги, а затем в сушильном шкафу при 105-110 °С до постоянной массы. Высушенный катионит хранят в плотно закрытом сосуде.
Для определения каталитической активности катионита измеряют его статистическую обменную емкость (СОЕ). Для этого в колбу с притертой пробкой емкостью 200 мл помещают навеску сухого катионита (1 г), взвешенную с точностью до 0,01 г, и добавляют 100 мл 0,1 н раствора гидроксида натрия. Колбу закрывают и оставляют стоять не менее 10 часов, периодически перемешивая ее содержимое встряхиванием.
Затем отбирают пипеткой 25 мл раствора и титруют 0,1 н раствором соляной кислоты в присутствии метилового оранжевого.
Рассчитывают СОЕ (в мэкв/л) по формуле:
СОЕ=n(VNk-V1 N1 k1)/g,
где: n - отношение общего объема анализируемого раствора к объему, взятому на анализ; V - объем раствора, взятого на анализ, мл; V1 - объем 0,1 н раствора НСl, пошедший на титрование, мл; N и N1 - нормальность растворов NаОН и НСl соответственно; k и k1 - поправочные коэффициенты к нормальностям растворов NаОН и НСl, соответственно; g - масса катионита, взятая на анализ, г.
СОЕ катионита должна быть в пределах 4,8 и 5,1 мэкв/г по гидроксиду натрия.
Техника безопасности
При выполнении лабораторной работы по алкилированию фенола особую опасность представляет фенол (карболовая кислота) – С6Н5ОН ММ = 94,11.
Физические свойства. Бесцветные кристаллы, краснеющие на воздухе, особенно на свету. С водой образует гидрат СбН5ОН × Н2О. Переходит в жидкое состояние уже при прибавлении незначительных количеств воды. Хорошо растворим в хлороформе, эфире, маслах. Коэффициент распределения масло/вода 4-10. Технический фенол - красно-бурая, иногда черная вязкая жидкость. Порог восприятия запаха 0,004 мг/л.
Возможны отравления парами фенола (особенно при его нагревании), мелкой пылью, образующейся из конденсирующихся паров, а главное - при попадании на кожу. Острые отравления происходят главным образом при попадании фенола на кожу. Тяжесть их зависит от размеров и степени поражения кожи и скорости оказания первой помощи.
Неотложная терапия. При смачивании одежды фенолом - немедленное удаление с работы, даже при кажущемся хорошем состоянии пострадавшего. Немедленно сменить одежду. Обтирание пораженных мест 10-40%-ным этиловым спиртом или растительными маслами. Обмывание всего тела водой с мылом (теплый душ). По показаниям: покой, согревание, ингаляции кислорода, искусственное дыхание, кофеин, камфара, кардиамин, внутривенно глюкоза (40% на физиологическом растворе), 30%-ный раствор тиосульфата натрия (8-10 мл). В случае раздражения слизистых оболочек верхних дыхательных путей -щелочные ингаляции. При (отравлении через рот дать выпить несколько стаканов теплой воды или взвеси жженой магнезии в воде (20: 200), вызвать рвоту (можно ввести под кожу 0,5-0,8 мл 1 %-ного раствора апоморфина). При необходимости - промывание желудка либо теплой водой с активированным углем, либо взвесью жженой магнезии, либо раствором сернокислого натрия до исчезновения запаха фенола. Позже - касторовое масло, яичный белок, слизистые отвары; глотать кусочки чистого льда Предельно допустимая концентрация 0,3 мг/м3.
Индивидуальная защита. Меры предупреждения. При наличии паров фенола -промышленный фильтрующий противога







